





- Sustainable biomaterials: hydrothermal synthesis of carbonated hydroxyapatite from buffalo bone waste
Jumiarti Andi Loloa,b, Endang Haryatia,c, Muhammad Arifind and Yusril Yusuf a,*
aDepartment of Physics, Faculty of Mathematics and Natural Science, Universitas Gadjah Mada, Yogyakarta 55281, Indonesia
bDepartment of Physics Education, Faculty of Teacher Training and Education, Universitas Kristen Indonesia Toraja, Tana Toraja 91811, Indonesia
cDepartment of Physics, Faculty of Mathematics and Natural Science, Universitas Cenderawasih, Jayapura 99351, IndonesiaThis article is an open access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
The synthesis of carbonated hydroxyapatite (CHA) from buffalo bone waste using the hydrothermal method offers a sustainable and cost-effective approach for producing biomimetic materials that closely resemble natural bone minerals. This study explores the impact of hydrothermal temperatures (120 °C, 160 °C, and 200 °C) on the crystallinity, phase composition, and morphology. Buffalo bone waste underwent chemical treatments, was calcined at 1000 °C, and subsequently used as a precursor for CHA. Characterization using X-ray diffraction (XRD), scanning electron microscopy with energy-dispersive X-ray spectroscopy (SEM-EDX), and Fourier-transform infrared spectroscopy (FTIR) revealed key findings, XRD analysis indicated that higher hydrothermal temperatures improve crystallinity, with sharper diffraction peaks at 200 °C, especially after 24-hour aging. SEM analysis showed morphological differences, with CHA synthesized at 200 °C exhibiting the densest, most uniform structure, whereas lower temperatures resulted in more porous structures with incomplete crystallization. FTIR spectra confirmed carbonate substitution in the hydroxyapatite lattice, while EDX analysis demonstrated a composition of calcium, phosphate, and trace elements, aligning with natural bone composition. This approach highlights CHA's potential as a biowaste-derived material for bone-related biomedical applications.
Keywords: Buffalo bone waste, Carbonated hydroxyapatite, Hydrothermal.
In recent years, the demand for sustainable biomaterials has increased, particularly in medical applications and tissue engineering. Hydroxyapatite (HA), a calcium phosphate compound with the chemical formula Ca₁₀(PO₄)₆(OH)₂, has garnered considerable interest in biomaterials and tissue engineering due to its excellent biocompatibility, osteoconductivity, and chemical similarity to the mineral component of natural bone and teeth [1–3]. These properties make hydroxyapatite an excellent choice for various biomedical applications, such as bone grafts, dental implants, drug delivery systems, and scaffold materials for bone tissue engineering [4–6]. HA supports cell adhesion, proliferation, and differentiation, along with its capacity to form direct bonds with natural bone, further strengthening its suitability for such applications.
Although synthetic hydroxyapatite is widely used, it often differs from natural bone mineral in composition, crystallinity, and morphology. Hydroxyapatite, while
exhibiting excellent biocompatibility and osteoconductivity, has inferior mechanical properties when compared to human bones, particularly its low resistance to fracture. For this reason, pure HA is often limited to applications that do not require excessive mechanical loading. To expand its use as a bone substitute, its mechanical properties can be significantly improved by controlling its density, microstructure, and through the incorporation of reinforcement phases, forming composites with ceramics and metallic materials that show promising mechanical properties for filling bone defects [7–9]. Natural bone is not composed of pure hydroxyapatite but rather a carbonated form of hydroxyapatite, where carbonate ions partially substitute for phosphate or hydroxyl groups in the crystal lattice. The presence of carbonate in the hydroxyapatite structure introduces lattice strain and structural defects, which, while slightly reducing the material's mechanical strength, enhances its biological performance by making it more resorbable and bioactive in physiological environments. The synthesis of carbonated hydroxyapatite (CHA) has become a topic of considerable interest, as it more closely mimics the composition and properties of natural bone than stoichiometric hydroxyapatite.
Several methods have been developed to synthesize CHA, including wet chemical precipitation, sol-gel processes, hydrothermal synthesis, and biomimetic approaches [3, 4, 10–13]. The hydrothermal method is valued for its ability to produce crystalline materials with specific shapes and high phase purity under mild conditions. The properties of hydroxyapatite can be refined by adjusting temperature, duration, pH, and precursor concentration [12, 14, 15]. One of the most sustainable approaches to producing hydroxyapatite, including its carbonated variant, is natural biowaste, such as animal bones. Research indicates that animal bones can serve as precursors for hydroxyapatite synthesis, suggesting the potential to convert this biowaste into valuable biomaterials. In recent years, marine organisms, including various fish bones and even putterfish teeth, have gained attention as alternative and sustainable sources for extracting natural HA, suitable for biomedical applications [8, 16–19]. Utilizing biogenic sources aligns with green chemistry principles by recycling waste materials and provides a more cost-effective alternative to synthetic routes. Buffalo bone is notable for its high calcium content and availability as a byproduct of the meat industry. Its potential as a precursor for hydroxyapatite synthesis further emphasizes the value of converting such biowaste into useful biomaterials [18, 20–22].
In the context of carbonated hydroxyapatite synthesis, buffalo bone waste offers an excellent starting material due to its inherent mineral composition, which already includes traces of carbonate, magnesium, sodium, and other elements in human bone. The process starts with collecting and preparing bones, which provides for cleaning, deproteinization, and calcination to eliminate organic materials and enhance the mineral phase [18, 21, 23, 24]. This mineral-rich phase can then be subjected to hydrothermal treatment to induce CHA formation, explicitly focusing on optimizing synthesis parameters to achieve desired structural and functional properties.
This study synthesizes carbonated hydroxyapatite from buffalo bone waste using a hydrothermal method, examining how different synthesis temperatures affect crystallinity, phase composition, and morphology. The objective is to identify the optimal temperature and aging time for producing CHA that closely mimics the composition and performance of natural bone minerals. CHA derived from buffalo bone waste has versatile biomedical applications beyond bone substitution, including dentistry, implant coatings, and drug delivery. Its integration into modern medicine supports sustainability and biocompatibility while addressing waste disposal challenges. By focusing on hydrothermal synthesis, this study contributes to biomaterials research by providing a sustainable method for producing a highly relevant bone substitute, supporting broader goals of resource efficiency and waste minimization. Through a detailed analysis of the synthesis process's structural, compositional, and morphological outcomes, the research aims to understand how biowaste can be transformed into high-performance biomaterials suitable for diverse medical applications.
Materials
Buffalo bone waste used as the Ca was collected from Tana Toraja, South Sulawesi, Indonesia. Diammonium hydrogen phosphate ((NH4)2HPO4) was used as a
phosphate source, and ammonium bicarbonate (NH4HCO3) as a carbonate source was purchased from Merck (USA).
Preparation of buffalo bone powder
Buffalo bone waste was collected and prepared as a precursor for synthesizing CHA. The preparation of buffalo bone begins with boiling to remove residual meat, followed by thorough cleaning under running water and sun-drying. The bones are cut into smaller pieces (approximately 1 cm) and subjected to sequential chemical treatments. Initially, they are immersed in a 0.1% NaOH solution for 24 h to eliminate lipids and enhance hydroxyl groups, then rinsed with deionized water. In the second stage, the bones are soaked in acetone for 24 h to remove any remaining organic matter and residual chemicals. The bones are then dried in an oven at 100 °C for 6 h, ground into fine powder using a ball mill, and calcined at 1000 °C for 5 h. The resulting powder is sieved through a 200-mesh sieve to achieve homogeneity in particle size. The powder was analyzed for its chemical composition and structure using XRD, FTIR, and SEM-EDX.
Synthesis of carbonated hydroxyapatite
Carbonated hydroxyapatite was synthesized by dissolving the calcined bone powder in 60 ml of deionized water using a magnetic stirrer set to 400 rpm for 1 hour to form a homogeneous Ca(OH)2 solution. Separately, (NH4)2HPO4 and NH4HCO3 are dissolved in deionized water and gradually combined to generate a carbonate-phosphate solution under basic pH conditions. This carbonate-phosphate solution is incrementally added to the Ca(OH)2 solution while stirring and heating to 60 °C for 60 minutes to facilitate the reaction. Upon achieving the desired pH, which was maintained in the range of pH 9-10, the mixture is transferred to a heat-resistant autoclave and subjected to hydrothermal treatment at varying temperatures of 200 °C, 160 °C, and 120 °C for 12 and 24 hours. After hydrothermal, the autoclave is allowed to cool naturally to room temperature. The samples are then precipitated by centrifugation and dried in an oven at 80 °C for 24 hours to obtain the final product.
Characterization of Materials
Morphological Analysis: Scanning Electron Microscopy (SEM) was used to observe the CHA sample surface, particle size distribution, and porosity. SEM provided high-resolution images that allowed for a more detailed examination of the density and uniformity of CHA particles, which are important for understanding the material's structure.
Functional Group Analysis: Fourier-transform Infrared Spectroscopy (FTIR) was employed to detect functional groups within CHA. FTIR identified the presence of phosphate (PO43-), hydroxyl (OH-), and carbonate (CO32-) groups, confirming the carbonate substitution in the hydroxyapatite structure. This analysis provided information about the chemical composition of CHA and its similarity to natural bone minerals.
Phase Analysis: X-ray Diffraction (XRD) was used to determine CHA's phase purity and crystallinity through its diffraction patterns. The XRD results offered insights into the lattice structure, crystal size, and degree of ordering within CHA, which are crucial for assessing its potential applications in the biomedical field.
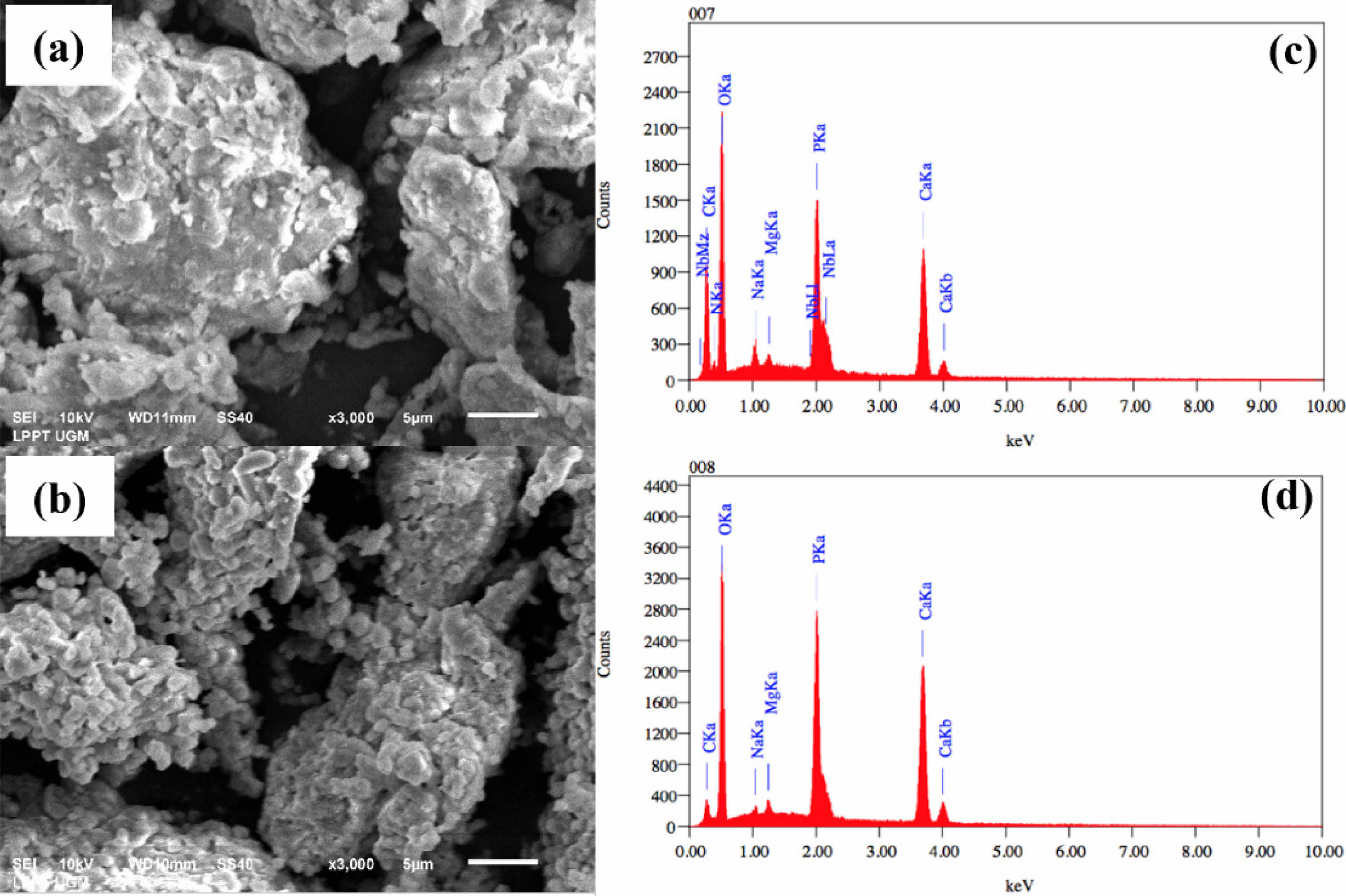
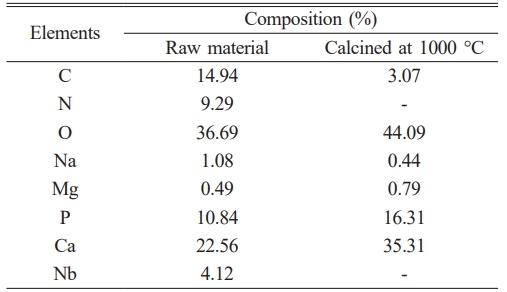
Figure 1(a-b), based on SEM result, shows that the particles in the sample are not spherical. Their arrangement and spacing are irregular, and they exhibit a range of sizes and crystalline forms. Fig. 1(c-d) presents the energy dispersive X-ray spectroscopy (EDX) test results, which are summarized in Table 1. The EDX test results indicate the components present in the material. In addition to calcium and phosphate, the main components of hydroxyapatite and other elements, such as magnesium and sodium, were also observed. Magnesium and sodium in the samples are inorganic elements commonly found in natural bone, specifically in buffalo and human bones. The wavenumber range from 4000 cm-1 to 1400 cm-1 is dedicated to identifying functional groups corresponding to stretching vibration absorptions for structural elucidation. The region beyond the 1400 cm-1 wavenumber is known as the fingerprint region, as each organic compound exhibits unique absorption characteristics in this area.
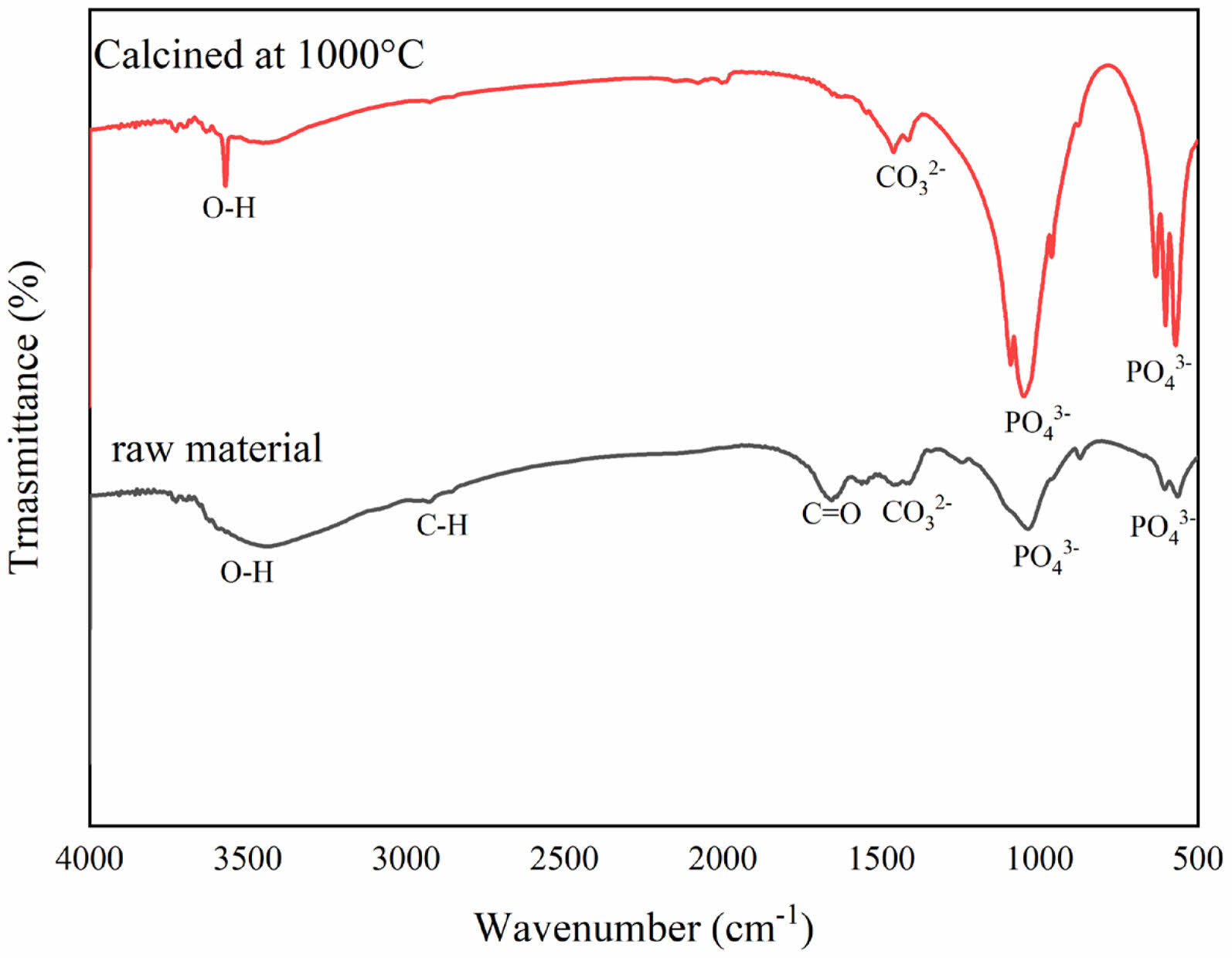
Figure 2 shows the FTIR spectrum of raw material and calcined at 1000 °C. Functional group analysis qualitatively identifies the components of buffalo bone through transmission absorption generated by FTIR. The raw material spectrum shows numerous and complex peaks, indicating the presence of various organic components within the bone.
The broad peak around 3000-3600 cm-1 coresspond to O-H bond stretching, which is associated with water molecules or hydroxyl groups in collagen or calcium phosphate hydroxide. The peak near 2940 cm-1 indicates C-H bond stretching, commonly found in lipids or other organic components. Additionally, the peak around 1650 cm-1 shows amide I (C=O) vibrations, characteristic of collagen protein [25]. After calcination, many of the peaks associated with organic components disappear or decrease in intensity, indicating the degradation of organic materials like collagen due to heat exposure. The sharper and more defined peaks in the calcined bone spectrum, particularly those around 1000-1100 cm-1, correspond to phosphate (PO4⁻) vibrations, which are the primary components of hydroxyapatite, the mineral phase of bone. The reduction or disappearance of amide peaks suggests the decomposition of collagen protein during the calcination process. High-temperature calcination causes the breakdown of organic matter in the bone, leaving only the heat-resistant inorganic components. After calcination, the remaining peaks in the bone powder spectrum reflect the crystalline structure of bone minerals, which become more prominent after the organic matter is removed. Also, peaks detected in the wavenumber range of 3300-3600 cm-1 indicate the presence of hydroxyl groups [26, 27]. Hydroxyapatite contains O-H, CO3-, and PO4⁻ groups. The phosphate group (PO4⁻) shows absorption bands that vibrate asymmetrically in the stretching mode at wavenumbers between 1028-1099 cm-1 without sintering and in the bending mode at wavenumbers 559 cm-1 and 601 cm-1 [4, 10, 18, 19].
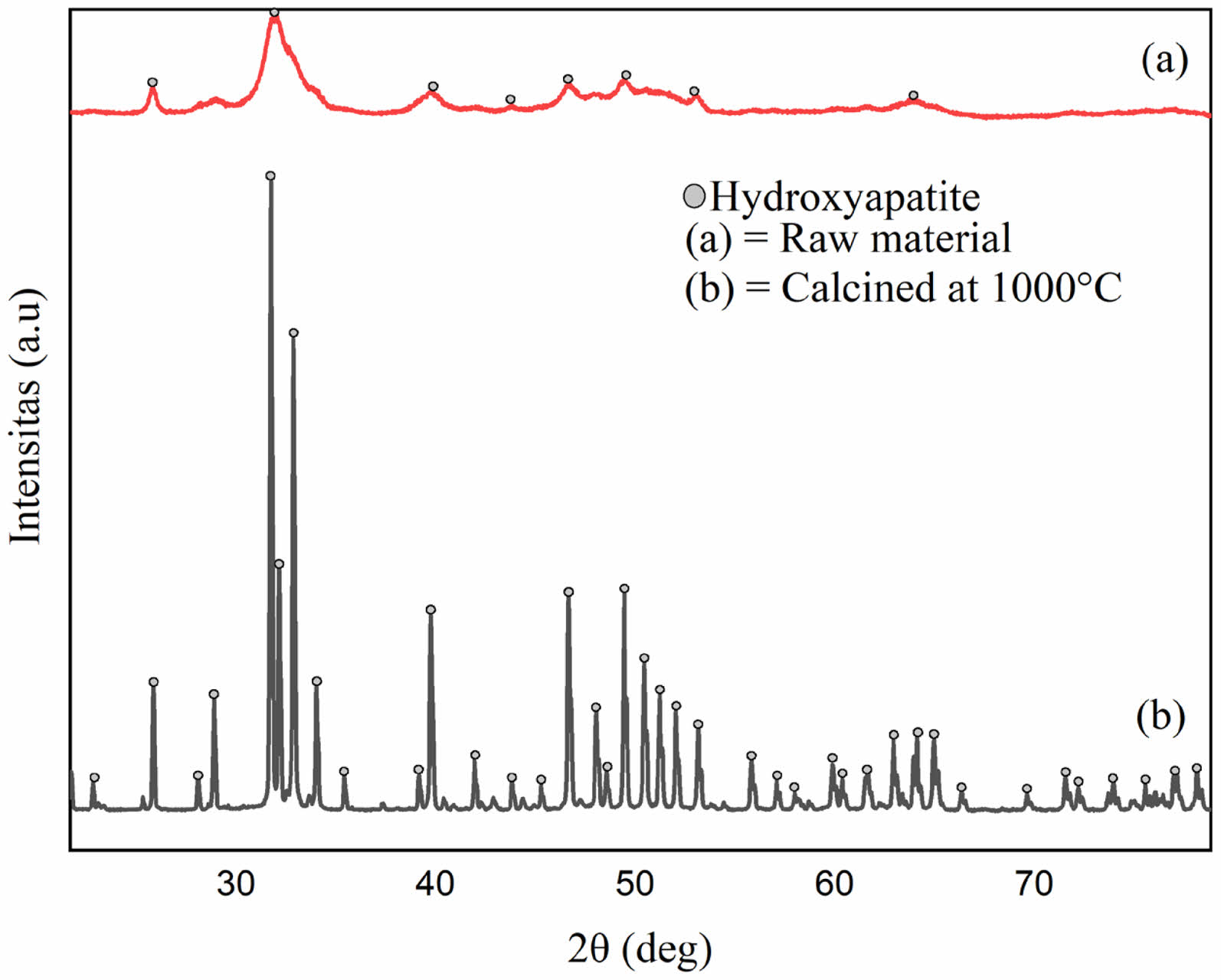
This finding aligns with the current study, where phosphate groups are detected at 1035 cm-1 for uncalcined bone powder and 1048 cm-1 for calcined bone powder. The presence of carbonate groups in the sample is not a negative feature, as human bones naturally contain carbonate elements that substitute for phosphate groups. Fig. 3 presents the X-ray diffraction patterns for raw material and material calcined at 1000 °C. Before calcination, the bone powder naturally contains hydroxyapatite within its mineral matrix.
The broad and low-intensity peaks indicate that the bone powder has a relatively amorphous structure or low crystallinity, typical of uncalcined bone, and often contains hydroxyapatite in a less organized, more complex form due to organic matter and water. Calcination involves heating the bone to high temperatures to eliminate organic material and water, leaving an inorganic calcium-rich structure. As shown in Fig. 3, the calcined bone powder displays sharper and more distinct peaks, indicating improved crystallinity of hydroxyapatite. This process removes or reduces organic components and water from the bone, resulting in a purer and more well-crystallized mineral phase.
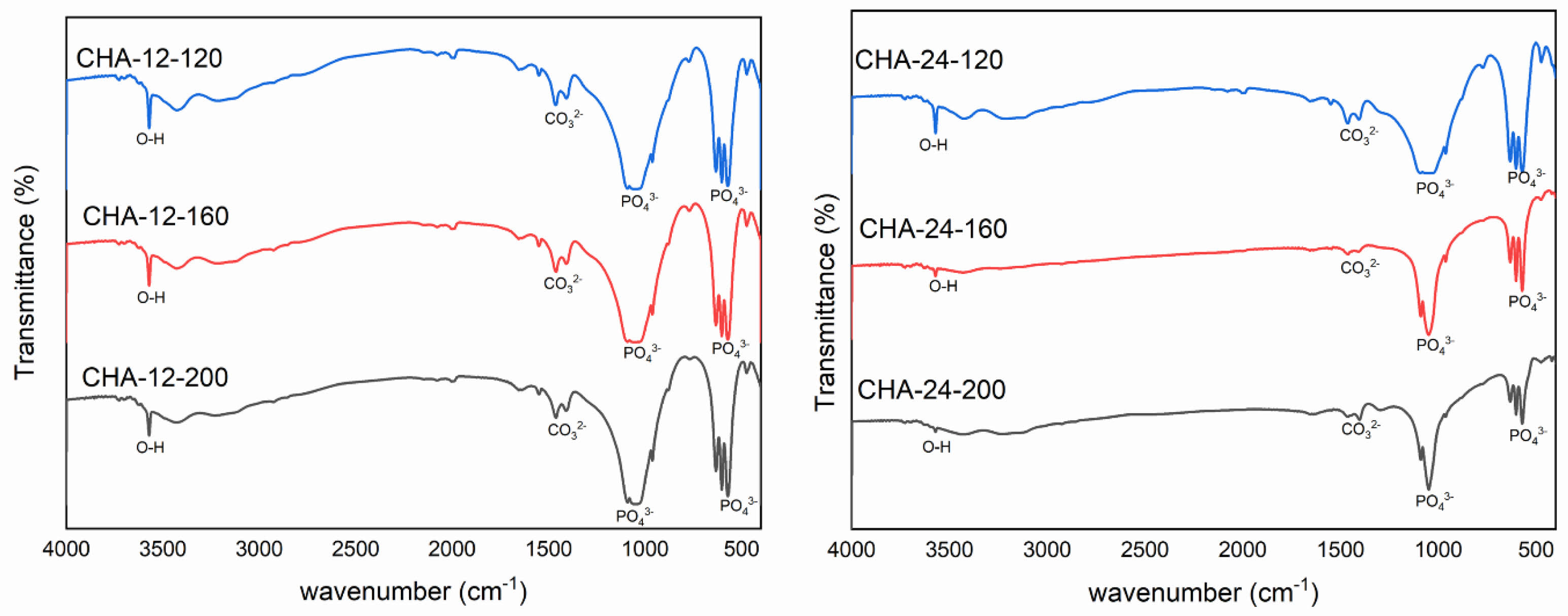
Figure 4(a) shows the FTIR spectra of carbonated hydroxyapatite (CHA) samples aged for 12 hours at different hydrothermal temperatures: 120 °C (CHA-12-120), 160 °C (CHA-12-160), and 200 °C (CHA-12-200). The FTIR spectra of CHA reveal characteristic peaks associated with specific functional groups. Carbonated hydroxyapatite, a modified form of hydroxyapatite (HA), which is the main mineral in bones and teeth, undergoes carbonation through the substitution of phosphate groups (PO43-) with carbonate groups (CO32-) within the HA crystal lattice [25, 28–31]. FTIR analysis of CHA reveals two typical types of carbonate substitution: type A, where carbonate (CO32-) replaces hydroxyl groups (OH-), and type B, where carbonate replaces phosphate groups (PO43-) [28, 32, 33]. Spectral characteristics of these substitutions can be identified at specific wavenumbers. Type A substitution carbonate peaks appear around 1540 cm-1 and 875 cm-1, while type B substitution peaks are observed around 1450 cm-1 for asymmetric stretching and 1415 cm-1 for symmetric stretching, along with a weak peak near 875 cm-1 [33–36]. Peaks around 3000-3600 cm-1 correspond to hydroxyl groups (OH). For CHA-12-120, hydroxyl groups are observed at 3430.58 cm-1 and 3571.08 cm-1; for CHA-12-160, at 3428.10 cm-1 and 3571.01 cm-1; and for CHA-12-200, at 3424.75 cm-1 and 3570.59 cm-1. The wavenumbers 1408.72–1460.70 cm-1 (CHA-12-120), 1407.84–1460.97 cm-1 (CHA-12-160), and 1407.66–1462.25 cm-1 (CHA-12-200) indicate the presence of carbonate groups (CO32-), confirming carbonate substitution within the HA lattice characteristic of carbonated HA. Peaks at wavenumbers 1049.23 cm-1 and 570.73–632.17 cm-1 (CHA-12-120), 1057.37 cm-1 and 602.40–632.23 cm-1 (CHA-12-160), and 1041.98 cm-1 and 602.28–632.19 cm-1 (CHA-12-200) denote phosphate groups (PO43-). Temperature variations induce minor shifts in the phosphate peak positions, indicating structural adjustments. The consistent presence of type B carbonate peaks across all temperatures suggests that, under these hydrothermal conditions and alkaline pH, the substitution of phosphate groups by carbonate ions is the dominant carbonation mechanism.
Figure 4b illustrates the FTIR spectra of CHA synthesized under hydrothermal conditions for 24 hours at 120 °C (CHA-24-120), 160 °C (CHA-24-160), and 200 °C (CHA-24-200). The presence of hydroxyl (OH) groups is confirmed by the peaks at 3571.07 cm-1 in all three samples. The intensity of these peaks reflects the quantity or presence of the hydroxyapatite phase, indicating the extent of the hydroxyl incorporation during synthesis. The spectra of CHA-24-120, CHA-24-160, and CHA-24-200 display similar peaks characteristic of type B carbonate substitution in CHA, with carbonate (CO32-) peaks observed at 1460.54 cm-1, 1460.64 cm-1, and 1462.22 cm-1, respectively. Prominent phosphate (PO43-) peaks are found at 1048.76 cm-1 (CHA-24-120), 1048.98 cm-1 (CHA-24-160), and 1041.83 cm-1 (CHA-24-200). The double peaks around 570–632 cm-1 show reduced intensity, suggesting changes in the structure or crystallinity of the material as the hydrothermal temperature increases. Similar to the 12 hour aging, type B substitution remains dominant, indicating that while temperature enhances overall crystal perfection, the primary mode of carbonate incorporation is maintained.
The FTIR analysis confirmed the presence of type B carbonate substitution in all CHA samples. Higher hydrothermal temperatures are known to favor type B substitution (carbonate replacing phosphate groups) over type A (carbonate replacing hydroxyl groups), as type B is thermodynamically more stable at elevated temperatures. Increasing temperature facilitates lattice rearrangement and enhances carbonate ion incorporation into phosphate sites.
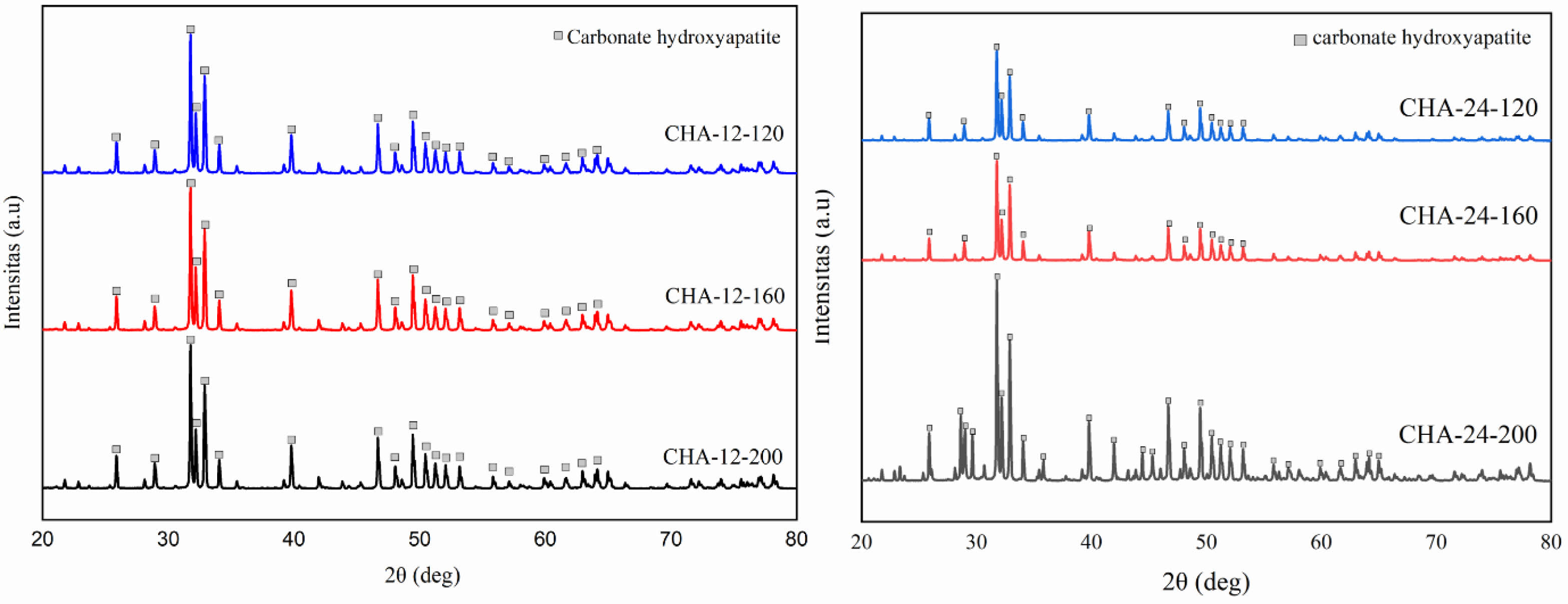
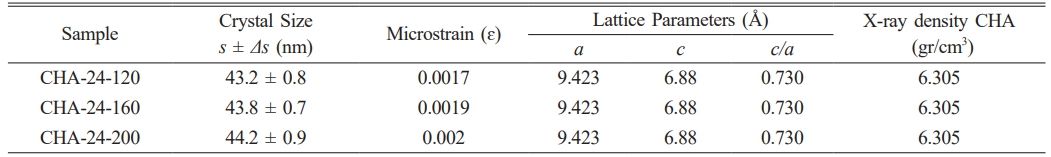
The X-ray diffraction (XRD) patterns of CHA samples aged for 12 hours are presented in Fig. 5a. Each peak in the diffraction pattern corresponds to the presence of a crystalline phase within the sample, with higher peak intensities indicating a higher concentration of the associated crystalline phase [3, 10, 37]. CHA-12-120 shows peaks at diffraction angles (2θ) of 31.77°, 32.19°, and 32.9° for the (112), (300), and (202) planes, respectively. CHA-12-160 has peaks at 31.76°, 32.18°, and 32.89°, while CHA-12-200 displays peaks at 31.77°, 32.19°, and 32.9° [2–4, 29].
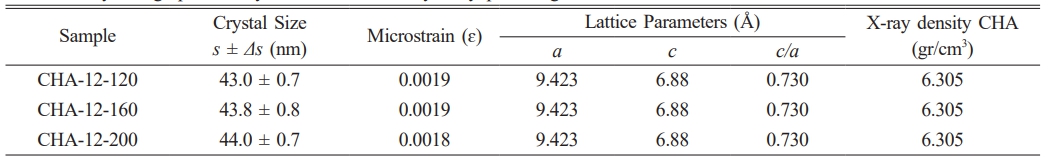
In all samples (CHA-12-120, CHA-12-160, and CHA-12-200), the marked peaks are consistent across different temperatures, indicating that the CHA phase is present and stable under all tested conditions. The variation in peak intensities among these samples suggests differences in crystallinity or the amount of phase formed at different temperatures. Sharper and more distinct peaks indicate higher crystallinity, while broader peaks suggest smaller crystallite sizes or the presence of distortions within the crystal structure. Crystallographic analysis of the carbonated hydroxyapatite aged for 12 hours is detailed in Table 2. Fig. 5b illustrates the XRD patterns of CHA samples aged 24 hours at various hydrothermal temperatures. Fig. 5b displays three characteristic peaks at diffraction angles of 31.77, 32.19, and 32.9 for the CHA-24-120 sample, 31.78, 32.2, and 32.91 for CHA-24-160, and 31.77, 32.19, and 32.9 for CHA-24-200. At 120 °C (CHA-24-120), the diffraction pattern shows lower intensity compared to the patterns at 160 °C (CHA-24-160) and 200 °C (CHA-24-200), indicating that crystallinity is reduced at 120 °C compared to the higher temperature samples. Many well-defined and sharp peaks reflect high crystallinity, as ordered crystalline structures produce more complex and distinct diffraction patterns. The crystallographic data of carbonated hydroxyapatite aged for 24 hours are summarized in Table 3. Crystallite size also influences peak intensities, where increased sharpness and peak intensity correlate with improved crystallinity and larger crystal sizes [37, 38].
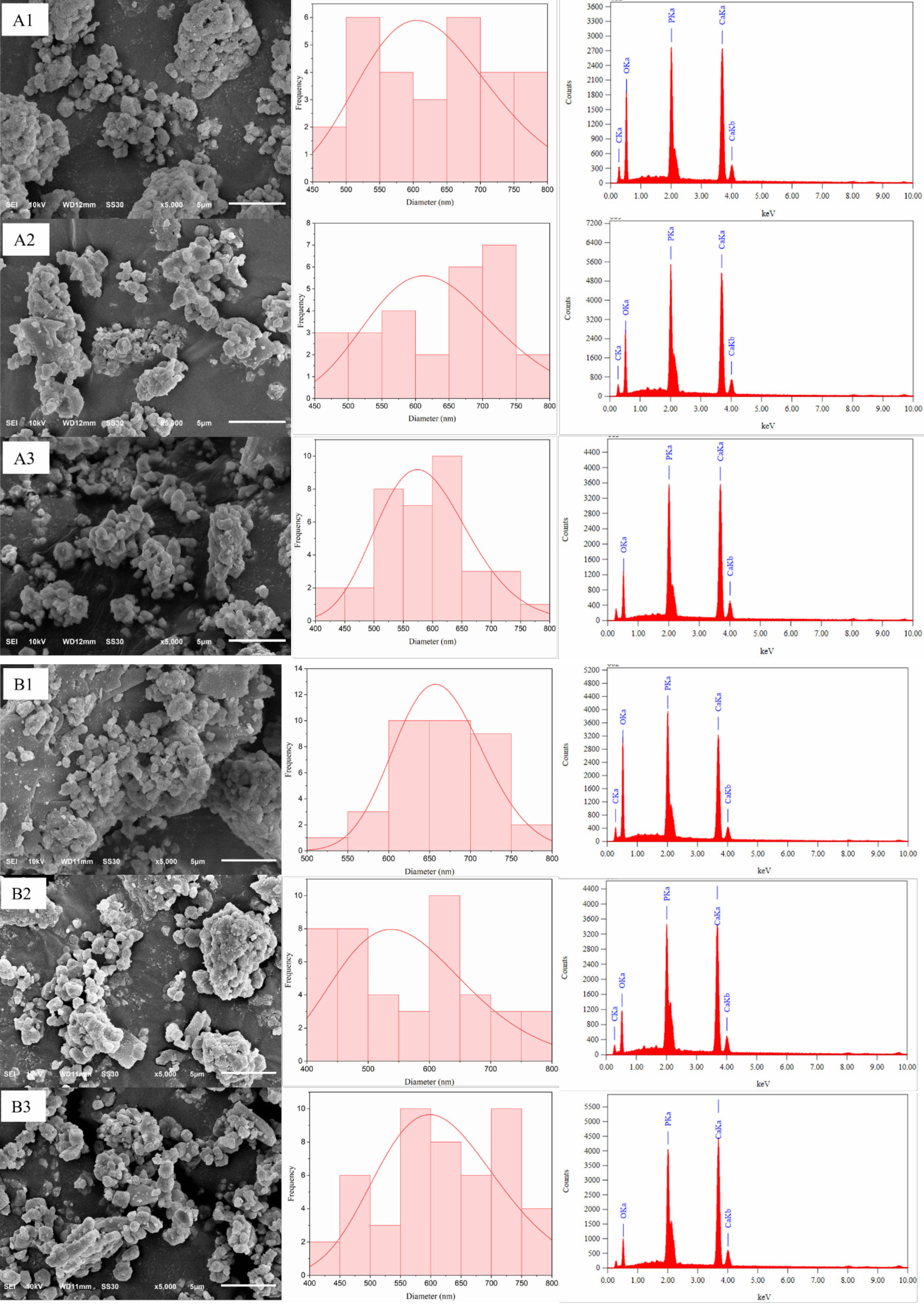
Figure 6a illustrates the surface morphology, particle distribution, and EDX analysis of carbonated hydroxyapatite (CHA) samples aged 12 hours at different hydrothermal temperatures: 120 °C, 160 °C, and 200 °C. The sample synthesized at 120 °C (CHA-12-120) displays a relatively rough morphology with large, irregular particles and significant porosity, characterized by numerous agglomerates. This suggests incomplete crystallization at this temperature, resulting in limited interparticle connectivity and a less dense structure. At 160 °C (CHA-12-160), the particles are more integrated and exhibit a more uniform shape, though the surface remains relatively rough with reduced pore size compared to CHA-12-120. The CHA-12-200 sample synthesized at 200 °C demonstrates the most uniform and dense morphology among the samples aged for 12 hours, with smoother particle surfaces, reduced porosity, and fewer agglomerates. This indicates significant crystal growth at 200 °C, enhancing particle interconnectivity and material density, which is expected to improve mechanical properties.
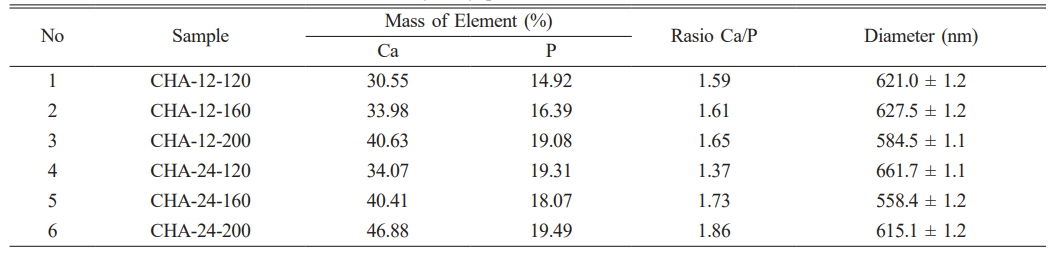
Figure 6b shows the morphology, particle distribution, and EDX analysis of CHA samples aged for 24 hours. The sample synthesized at 120 °C (CHA-24-120) has a heterogeneous surface with grains and clusters of small to medium sizes, along with noticeable agglomerates. The grains are irregular in shape, indicating incomplete or unstable crystallization at this lower temperature. At 160 °C (CHA-24-160), the surface morphology is smoother and more uniform than the sample synthesized at 120 °C, with more homogeneous grain sizes and fewer large agglomerates. This suggests enhanced crystallization and grain growth at 160 °C, as grains exhibit a more rounded and regular morphology, indicative of improved crystal quality. The CHA-24-200 sample synthesized at 200 °C displays the most uniform and compact morphology among the three samples, with a very smooth surface, more minor and uniformly distributed grains, and a high degree of homogeneity, reflecting optimal crystallization and material densification at this elevated temperature. Table 4 presents the particle size of the samples and the EDX analysis results, which will be used to calculate the Ca/P ratio of carbonated hydroxyapatite. The table indicates that the sample closest to the bone ratio is CHA-24-160, with a Ca/P ratio of 1.73 [2, 38, 39]. This value is remarkably close to the Ca/P ratio found in natural bone, which typically ranges from 1.67-1.71. This optimal Ca/P ratio has significant implications for its biological performance. A material with a Ca/P ratio closely mimicking natural bone is expected to exhibit superior biocompatibility, as it is more readily recognized and integrated by the host physiological system. Furthermore, such a ratio is crucial for enhanced osteoconductivity, facilitating the direct growth of new bone onto the materials surface. Therefore, the CHA-24-160 sample is considered the most biomimetically suitable composition among the synthesized materials.
|
Fig. 1 Morphology (a-b) and composition distribution (c-d) for raw material and calcined at 1000 °C. |
|
Fig. 2 FTIR Spectrum of raw material and calcined at 1000 °C. |
|
Fig. 3 X-ray Diffraction Patterns of (a) raw material and (b) calcined at 1000 °C. |
|
Fig. 4 FTIR spectrum of carbonated hydroxyapatite (CHA) with (a) 12 hours of aging and (b) 24 hours of aging. |
|
Fig. 5 X-ray Diffraction Patterns of CHA with (a) 12 hours of aging and (b) 24 hours of aging. |
|
Fig. 6 Morphology, particle distribution, and EDX analysis of (A1) CHA-12-120, (A2) CHA-12-160 and (A3) CHA-12-200 with 12 hours of aging. |
|
Table 2 Crystallographic Analysis of Carbonated Hydroxyapatite Aged for 12 Hours. |
|
Table 3 Crystallographic Analysis of Carbonated Hydroxyapatite Aged for 24 Hours. |
This study explores the synthesis of carbonated hydroxyapatite (CHA) from buffalo bone waste using the hydrothermal method with varying temperature settings. The findings indicate that hydrothermal synthesis parameters, particularly temperature and aging duration, significantly influence CHA's crystallinity, phase purity, and morphology. The consistent control of pH (maintained in the range of pH 9-10) during the hydrothermal process was crucial for successful CHA formation. Higher hydrothermal temperatures (160 °C and 200 °C) notably enhanced CHA's crystallinity and morphological uniformity, as evidenced by sharper and more distinct X-ray diffraction peaks and a denser particle morphology observed through scanning electron microscopy (SEM). FTIR analysis confirmed the incorporation of type B carbonate substitutions in the hydroxyapatite lattice, closely mimicking the mineral composition of natural bone. The consistent presence of type B carbonate peaks across all temperature suggest that, under these hydrothermal conditions and alkaline pH, the substitutions of phosphate groups by carbonate ions is the dominant carbonation mechanism. Higher hydrothermal temperatures are known to favor type B substitution (carbonate replacing phosphate groups) over type A (carbonate replacing hydroxyl groups), as type B is thermodynamically more stable at elevated temperatures. Increasing temperature facilitates lattice rearrangement and enhances carbonate ion incorporation into phosphate sites. Furthermore, EDX analysis revealed that the CHA-24-160 sample exhibited a Ca/P ratio of 1.73, remarkably close to the 1.67-1.71 ratio found in natural bone. This optimal Ca/P ratio has significant implications for its biological performance, enhancing biocompatibility and osteoconductivity. FTIR and EDX also confirmed that the successful removal of organic components during calcination underscores the transformation from a composite organic-inorganic matrix to a predominantly inorganic and highly crystallized structure. These improvements suggest that CHA synthesized from buffalo bone waste exhibits promising characteristics for applications in bone tissue engineering and other biomedical fields due to its superior structural and compositional attributes. Future research should focus on optimizing synthesis conditions and conducting rigorous in vitro and in vivo assessments to fully establish CHA’s efficacy as a bone substitute or scaffold material in clinical applications.
The authors gratefully acknowledge financial support from Indonesian Education Scholarship (BPI), Center for Higher Education Funding and Assessment Ministry of Higher Education, Science, and Technology of Republic Indonesia, and Indonesia Endowment Fund for Education (LPDP) for this research (00843/J5.2.3./BPI.06/9/2022). The research was also financially supported by Directorate of Research Universitas Gadjah Mada through the RTA Program (4193/UN1.P1/Dit-Lit/PT.01.03/2025). The authors also acknowledge the support of LPPT UGM, Indonesia for providing research facilities.
- 1. S.E. Cahyaningrum, N. Herdyastuty, B. Devina, and D. Supangat, in IOP Conf. Ser. Mater. Sci. Eng (Institute of Physics Publishing, 2018) pp.1-5.
-
- 2. M. Sari, Aminatun, T. Suciati, Y.W. Sari, and Y. Yusuf, Coatings 11[10] (2021) 1-15.
-
- 3. R. Wati and Y. Yusuf, in IOP Conf. Ser. Mater. Sci. Eng (Institute of Physics Publishing, 2019) pp.1-7.
-
- 4. I.K. Januariyasa, I.D. Ana, and Y. Yusuf, Mater. Sci. Eng. C. 107 (2020) 1-13.
-
- 5. G. Thrivikraman, A. Athirasala, C. Twohig, S.K. Boda, and L.E. Bertassoni, Dent. Clin. North. Am. 61[4] (2017) 835-856.
-
- 6. O.A. Osuchukwu, A. Salihi, I. Abdullahi, B. Abdulkareem, and C. S. Nwannenna, SN Appl. Sci. 3[10] (2021) 822-844.
-
- 7. L.A.F. Vieira, I.B. da C.J. Meireles, and E.M.B. Sousa, J. Ceram. Process. Res. 23[5] (2022) 725-736.
-
- 8. S.A. Doğdu, C. Turan, T. Depci, and D. Ayas, J. Ceram. Process. Res. 22[3] (2021) 356-361.
-
- 9. N. Ekren, J. Ceram. Process. Res. 18[1] (2017) 64-68.
-
- 10. H.A. Permatasari and Y. Yusuf, in IOP Conf. Ser. Mater. Sci. Eng (Institute of Physics Publishing, 2019) pp.1-7.
-
- 11. J. Lamsihar Manalu, B. Soegijono, and D.J. Indrani, Asian J. Appl. Sci. 3[4] (2015) 758-765.
- 12. P. Szterner and M. Biernat, Bioinorg. Chem. Appl. 147[23] (2022) 1-13.
-
- 13. A. Yusuf, N.M. Muhammad, A.R. Noviyanti, and Risdiana, in Key Eng. Mater. (Trans Tech Publications Ltd, 2020) pp. 228-233.
-
- 14. C.I. Codrea, D. Lincu, I. Atkinson, D.C. Culita, A.M. Croitoru, G. Dolete, R. Trusca, B.S. Vasile, M.S. Stan, D. Ficai, and A. Ficai, Materials 17[7] (2024) 1-23.
-
- 15. J. Partini, S.M. Bilqis, F.A. Salimy, I. Aziz, M. Sari, N. Cahyati, and Y. Yusuf, J. Ceram. Process. Res. 25[4] (2024) 599-606.
-
- 16. S.A. Doğdu, C. Turan, T. Depci, E. Bahçeci, K. Sangün, and D. Ayas, J. Ceram. Process. Res. 25[1] (2024) 85-91.
-
- 17. K.W. Goh, Y.H. Wong, R.S.K. Singh, H. Chandran, S.K. Wong, and K.Y.S. Lee, J. Ceram. Process. Res. 23[2] (2022) 158-164.
-
- 18. I. Pawarangan and Y. Yusuf, in IOP Conf. Ser. Mater. Sci. Eng (Institute of Physics Publishing, 2018) pp.1-7.
-
- 19. J.A. Lolo, D.P.P. Ambali, W. Jefriyanto, D. Handayani, W. Afridah, E. A. Wikurendra, R. Amalia, and A. Syafiuddin, Biointerface Res. Appl. Chem. 12[2] (2022) 2440-2449.
-
- 20. J. Brzezińska-Miecznik, K. Haberko, M. Sitarz, M.M. Bućko, and B. Macherzyńska, Ceram. Int. 41[3] (2015) 4841-4846.
-
- 21. A. Ayadi and J. Bouaziz, Jom 74[8] (2022) 3119-3132.
-
- 22. I. Atemni, R. Ouafi, K. Hjouji, I. Mehdaoui, A. Ainane, T. Ainane, M. Taleb, and Z. Rais, Emergent Mater. 6[2] (2023) 551-560.
-
- 23. A. Pal, S. Paul, A.R. Choudhury, V.K. Balla, M. Das, and A. Sinha, Mater. Lett. 203 (2017) 89-92.
-
- 24. P. Surya, A. Nithin, A. Sundaramanickam, and M. Sathish, J. Mech. Behav. Biomed. Mater. 119 (2021) 1-9.
-
- 25. A.A. Shaltout, M.A. Allam, and M.A. Moharram, Spectrochim. Acta, Part A 83[1] (2011) 56-60.
-
- 26. J. Venkatesan, B. Lowe, P. Manivasagan, K.H. Kang, E. P. Chalisserry, S. Anil, D.G. Kim, and S.K. Kim, Materials 8[8] (2015) 5426-5439.
-
- 27. N. Cahyati, M. Sari, and Y. Yusuf, Key Eng. Mater. 977 (2024) 103-108.
-
- 28. W. Bonfield and I.R. Gibson, J. Biomed. Mater. Res. 59[4] (2002) 697-708.
-
- 29. M.A. Irfa’i, W.W. Schmahl, Y.M. Pusparizkita, S. Muryanto, A. Prihanto, R. Ismail, J. Jamari, and A.P. Bayuseno, J. Mol. Struct. 1306 (2024) 1-11.
-
- 30. M. Kamitakahara, T. Nagamori, T. Yokoi, and K. Ioku, J. Asian Ceram. Soc. 3[3] (2015) 287-291.
-
- 31. T. Kono, T. Sakae, H. Nakada, T. Kaneda, and H. Okada, Minerals 12[2] (2022) 1-11.
-
- 32. H. Copete, E. López, and C. Baudin, Bol. Soc. Esp. Ceram. Vidrio (2024) 1-13.
- 33. S.A. Siddiqi and U. Azhar, in Handbook of Ionic Substituted Hydroxyapatites (Elsevier, 2019), pp. 149-173.
-
- 34. M.E. Fleet and X. Liu, J. Solid State Chem. 174[2] (2003) 412-417.
-
- 35. K. Benataya, M. Lakrat, L.L. Elansari, and E. Mejdoubi, Mater. Today: Proc. 31 (2020) S83-S88.
-
- 36. M.R. Senra, R.B. de Lima, D. de H.S. Souza, M. de F.V. Marques, and S.N. Monteiro, J. Mater. Res. Technol. 9[4] (2020) 7190-7200.
-
- 37. M. Sari, N.A. Kristianto, Chotimah, I.D. Ana, and Y. Yusuf, Coatings 11[10] (2021) 1-18.
-
- 38. M.R. Habiburrohman, M.A. Jamilludin, N. Cahyati, N. Herdianto, and Y. Yusuf, RSC Adv. 15[7] (2025) 5135-5150.
-
- 39. M. Sari, P. Hening, Chotimah, I.D. Ana, and Y. Yusuf, Mater. Today Commun. 26 (2021) 1-12.
-
 This Article
This Article
-
2025; 26(5): 843-851
Published on Oct 31, 2025
- 10.36410/jcpr.2025.26.5.843
- Received on May 16, 2025
- Revised on Jul 19, 2025
- Accepted on Aug 29, 2025
Services
- Abstract
introduction
materials and methods
result and discussion
conclusion
- Acknowledgements
- References
- Full Text PDF
Shared
Correspondence to
- Yusril Yusuf
-
Department of Physics, Faculty of Mathematics and Natural Science, Universitas Gadjah Mada, Yogyakarta 55281, Indonesia
Tel : +62-0274-6492383 Fax: +62-0274-6492383 - E-mail: yusril@ugm.ac.id
Clean-Energy Research Institute(CRI), Hanyang University, 222, Wangsimni-ro, Seongdong-gu, Seoul, 04763, Korea
E-mail: jcpr@hanyang.ac.kr