





- Morphology and size control of 513 MHSH, Mg(OH)2 from sea bittern using H2SO4 and NH4OH
Jiyeon Kima,b, HyunSeung Shima, YeongJo Yuna, Seong-Ju Hwangb and YooJin Kima,*
aEngineering Materials Center, Korea Institute of Ceramic Engineering and Technology, Icheon 17303, Republic of Korea
bDepartment of Battery Engineering, Yonsei University, Seoul 03722, Republic of KoreaThis article is an open access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
513 MHSH (Magnesium Hydroxide Sulfate Hydrate) and Mg(OH)2 were synthesized using a domestic resource, sea bittern, as a Mg precursor instead of conventional reagents. Sea bittern contains a large amount of Ca2+ ions, which can be efficiently removed using sulfuric acid. We synthesized them using the obtained MgSO4 solution from sea bittern. By adding ammonia to the MgSO4 solution to synthesize plate-like Mg(OH)2 and adjusting the Mg(OH)2/MgSO4 ratio by adjusting the amount of ammonia, needle-like 513 MHSH were synthesized. In addition, the synthesized hexagonal plate-like Mg(OH)2 was grown to a size of 50 to 400 nm by hydrothermal reaction. The conditions for synthesizing the needle-like 513 MHSH were found by the addition of sulfuric acid. The synthesized 513 MHSH and Mg(OH)2 were characterized by X-ray Diffraction (XRD), Scanning Electron Microscopy (SEM) and Transmission Electron Microscopy (TEM).
Keywords: Sea bittern, Mg(OH)₂, Magnesium, CaSO4, MgSO4 solution.
Magnesium hydroxide sulfate hydrate [abbreviated MHSH, xMg(OH)2·yMgSO4·zH2O (x-y-z phase;)] has various structures depending on the ratio of Mg(OH)2, MgSO4 and H2O [1-3]. MHSH whiskers exist in various forms such as 513 MHSH (5Mg(OH)2·MgSO4·3H2O), 517 MHSH (5Mg(OH)2·MgSO4·7H2O), 512 MHSH (5Mg(OH)2·MgSO4· 2H2O) and 212 MHSH (2Mg(OH)2·MgSO4·2H2O) [4-8]. 513 MHSH whiskers, a needle-like inorganic material, have attracted attention due to their practical applicability, including their use in resins, fillers and fibrous composites and as a reinforcement material [9-12]. Due to the SO42- ion-bonded structure between the Mg(OH)2 layered structures, 513 MHSH can be used as a flame retardant because of the release of water molecules at temperatures higher than the usual dehydration temperature under fire conditions. 513 MHSH forms a structure in which layered Mg(OH)2 are bonded by SO42- ions, and the bonding with sulfate ions causes water molecules to be released at a higher temperature than the general dehydration temperature in fire conditions [13-16].
513 MHSH whiskers can be obtained by using various starting materials, such as MgO, MgSO4, MgCl2, or Mg(OH)2 [17-19]. 513 MHSH synthesis is possible by adjusting the MgSO4/MgO ratio through a hydrothermal reaction [20-22]. Pure 513 MHSH can be obtained with MgSO4 and ammonia, but the use of a strong base, such as NaOH, easily forms Mg(OH)2 and interferes with the formation of the 513 MHSH [23]. Furthermore, because Mg(OH)2 and MgSO4 are easily modified under acidic/basic conditions, 513 MHSH can be synthesized using Mg(OH)2 precursors and sulfuric acid [3, 24]. Our group previously investigated the synthesis and morphologies of 513 MHSH by adjusting the ratio of the reagent precursor MgO/MgSO4 and applied the results to find the conditions for synthesizing 513 MHSH using acid and base catalysts, H2SO4 and ammonia [3, 21-24].
Sea bittern removes Na+ ions from seawater through an ion exchange membrane, and Mg2+ ions are present at relatively high concentrations, so it is easy to extract Mg2+ ions from sea bittern [25]. However, because sea bittern contains Ca2+ ions at a higher concentration compared to seawater, Ca2+ ions must be removed efficiently to synthesize high-purity magnesium compounds [25]. Adding sulfuric acid to sea bittern can remove Ca2+ ions by taking advantage of the solubility difference between MgSO4 and CaSO4. Because the relatively low-soluble calcium sulfate (CaSO4, 2.55 g/L) is precipitated and the highly soluble magnesium sulfate (MgSO4, 351 g/L) remains as ions in the solution, the two substances can be easily separated. After the removal of the Ca2+ ions from the sea bittern using sulfuric acid, needle-like 513 MHSH and plate-like Mg(OH)2 were synthesized by adjusting the amount of basic precipitant in the purified MgSO4 solution.
Materials
We used sea bittern (Hanju Salt, Korea) as the Mg precursor. Sodium hydroxide (NaOH, Daejung Chem., Korea) and sulfuric acid (10% H2SO4, Daejung Chem., Korea) were used to extract the MgSO4 and Mg(OH)2. Sulfuric acid (95% H2SO4, Samchun Chem., Korea) and ammonium hydroxide (28% NH4OH, Daejung Chem., Korea) were used without further purification as the catalyst in the synthesis process for the 513 MHSH. An aqueous solution of the precursor systems was prepared with the appropriate quantities of reagents in distilled water, using various synthesis conditions to obtain the 513 MHSH whiskers. Then, the morphology and structure were examined by Scanning Electron Microscopy (SEM, Model JSM-6390, JEOL, Japan), Transmission Electron Microscopy (TEM, Model CM 200, Philips, Netherlands) and powder X-Ray Diffraction (XRD, Model D/Max 2500, Rigaku, Japan). Inductively Coupled Plasma Optical Emission Spectrometry (ICP-OES, Model SPECTRO ARCOS, SPECTRO Analytical Instruments, Germany) and X-ray Fluorescence (XRF, Model S1 TITAN, Bruker, USA) were used to identify the components of the raw materials and the solution.
Synthesis of the 513 MHSH
In general, 513 MHSH is synthesized using a hydrothermal method. Previously, we synthesized 513 MHSH adjusting the molar ratio of the MgSO4 and MgO, using the reaction of the single precursor MgSO4 and NH4OH (basic catalyst) and the reaction with Mg(OH)2 and H2SO4 (acid catalyst). These experiments utilized the reagents MgO and MgSO4 to synthesize high-purity 513 MHSH by adjusting the pH, using ammonia and sulfuric acid as catalysts. We investigated the effect of pH, a critical factor in 513MHSH synthesis, and selected NH₄OH, a weak base with better pH control than NaOH because elevated pH levels favored the formation of Mg(OH)₂ over 513 MHSH [21, 23].
It is difficult to produce uniform 513 MHSH whiskers because plate-like Mg(OH)2 particles were formed in high concentrations of OH- due to the interaction between Mg2+ and OH- [26, 27]. The amount of SO42- ions is also an important factor in whisker growth because it affects the one-dimensional growth of MHSH, and if the amount of SO42- ions is too small, Mg2+ ions combine with OH- ions rather than SO42- ions to form Mg(OH)2 instead of 513 MHSH [22].
In this work, we prepared MgSO4 and Mg(OH)2 using sea bittern as the Mg precursor instead of the reagent MgSO4 and Mg(OH)2 and synthesized high-quality 513 MHSH whiskers using NH4OH and H2SO4 as the catalyst to control the pH in two ways. In the first method, sulfuric acid was added to the sea bittern to form a MgSO4 solution, and NH4OH (basic catalyst) was added to the solution to adjust the pH conditions to synthesize the 513 MHSH. In the second method, 513 MHSH was synthesized by adding sulfuric acid as a catalyst to Mg(OH)₂, which was prepared by adding excess base to the MgSO₄ solution derived from the sea bittern. Sea bittern is concentrated seawater and is rich in Mg2+ ions but also contains high levels of other metal ions, including Ca²⁺ ions, due to the concentration process. Because the presence of Ca2+ ions interrupts the synthesis of high-purity Mg compounds, Ca2+ removal from the sea bittern is necessary. We added sulfuric acid and removed them by precipitation in the form of calcium sulfate (CaSO4). Calcium sulfate (CaSO4) is precipitated due to its low solubility, while MgSO4 remains dissolved because of its high solubility [28].
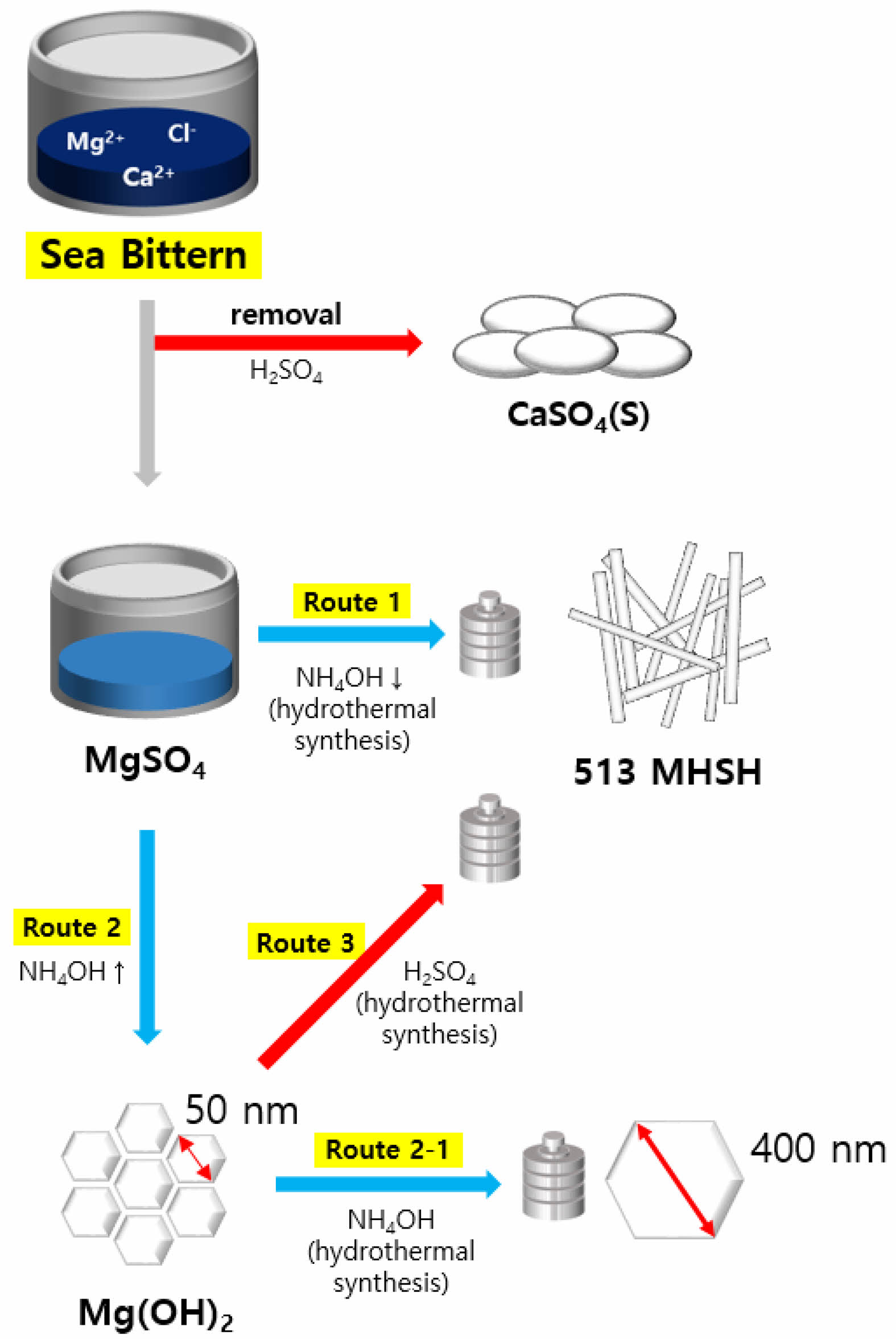
Figure 1 presents a schematic diagram showing the synthesis process of 513 MHSH from sea bittern using basic and acidic catalysts, including the Ca2+ removal process by adding sulfuric acid and the shape and size control through pH adjustment. In Route 1, after removing the Ca2+ ions with sulfuric acid, an appropriate amount of NH4OH was added to the MgSO4 solution to synthesize the 513 MHSH through hydrothermal synthesis. In Route 2, an excess amount of NH4OH was added to the MgSO4 solution at room temperature to obtain 50 nm hexagonal magnesium hydroxide. The particle size increases from 50 to 400 nm when hydrothermally synthesized in a basic condition in Route 3. We synthesized 513 MHSH using a hydrothermal reaction with a sulfuric acid catalyst in Route 3 and magnesium hydroxide in Route 2.
This study was done in three steps: 1) the removal process of Ca2+ from sea bittern using sulfuric acid to prepare a MgSO4 solution, 2) the synthesis process of 513 MHSH and Mg(OH)2 by adjusting the amount of basic precipitant, and 3) the synthesis process of 513 MHSH and Mg(OH)2 by adjusting the pH of the synthesized Mg(OH)2 solution. In the Ca2+ removal process, Ca2+ ions in the sea bittern are precipitated as CaSO4 by reacting sulfuric acid with the sea bittern. The 513 MHSH synthesis can be synthesized using two catalysts. The detailed experimental conditions for the synthesis of 513 MHSH are summarized in Table 1.
Extraction of MgSO4 using sulfuric acid
Adding sulfuric acid to sea bittern removes Ca2+ ions and creates a MgSO4 solution.
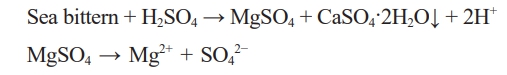
190 g of 10% H2SO4 were added to 100 g of sea bittern and vigorously stirred for 30 minutes. Adding an excess amount of sulfuric acid to sea bittern ensured sufficient SO42- ions in the form of MgSO4. After stirring, the precipitated CaSO4 was separated from the MgSO4 solution via centrifugation. This method involves removing Ca2+ ions by reacting them with SO42- ions to form calcium sulfate (CaSO4) through precipitation. Because MgSO4 exists as ions, it can act as a good precursor for the synthesis of Mg-based materials. In addition, because the ratio and amount of MgSO4 /MgO are important, the remaining MgSO4 solution was heated at 100℃ for 2 hours to evaporate 30% of the solution to control the molar concentration of MgSO4 solution based on previous experiments [20-22].
Synthesis of 513 MHSH using a basic catalyst and MgSO4 solution from sea bittern
When ammonia was added to the MgSO4 solution extracted from the sea bittern, some of the Mg2+ ions reacted with the OH- ions and were converted to Mg(OH)2. Mg(OH)2 combined with SO42− ions caused the formation of 513 MHSH.
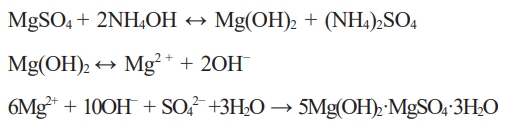
NH4OH (4 M) solution (65-80 mL) was added to the MgSO4 solution prepared from sea bittern at room temperature to produce the whiskers. In the subsequent hydrothermal procedure, the solution was transferred to a Teflon-lined autoclave with a 100 mL capacity. The autoclave was maintained at 180 °C for 48 h in a drying oven and then naturally cooled down to room temperature. The obtained products were centrifuged, washed with deionized water and dried in air at 80 °C for one day. Based on the previous studies that increased Mg²⁺ selectivity facilitates 513 MHSH formation, we did the experiment by adjusting the concentration of the MgSO4 solution obtained from the sea bittern [20-22]. To increase the Mg²⁺ concentration, 46-70 mL of 4 M NH₄OH were added after approximately 30% of the MgSO₄ solution had been evaporated, given its critical role in 513 MHSH synthesis.
Synthesis of Mg(OH)2 plates from sea bittern
Mg(OH)2 was synthesized by adding ammonia to MgSO4 obtained from the sea bittern under room temperature and pressure conditions. When a basic precipitant was added to the generated MgSO4 solution, OH- ions combined with the Mg2+ ions in the sea bittern to precipitate in the form of Mg(OH)2.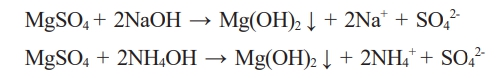
For the synthesis of Mg(OH)2, De-Ca sea bittern was added at a molar ratio of 1:2 to a basic precipitant, NaOH or NH4OH, and sufficiently stirred at 300 rpm for 2 hours. The obtained products were centrifuged at 8000 rpm for 10 minutes, washed with deionized water and dried in air at 80 °C for one day to obtain Mg(OH)2 as a powder [25].
Formation of Mg(OH)2 plate particles up to 400 nm
First, 0.5 g of Mg(OH)2 were dissolved in 10 mL of 2M NH4OH deionized water and then ultra-sonicated for 5 minutes. Then, the solution was transferred to a 25 mL capacity Teflon-lined autoclave. The autoclave was maintained at 180 °C for 1-24 hours in a drying oven and then allowed to cool naturally to room temperature. The obtained products were centrifuged, washed with deionized water and dried in air at 60 °C for one day.
Synthesis of 513 MHSH using a sulfuric acid catalyst and Mg(OH)2 from sea bittern
Mg(OH)2 at the desired concentration (4 M) was dissolved in 50 mL of deionized water and subjected to ultra-sonication for 5 min. After 10 to 30 mL of 5 M H2SO4 solution was added, the solutions were transferred to a 100 mL capacity Teflon-lined autoclave. The autoclave was maintained at 180 °C for 48 hours in a drying oven and then allowed to cool naturally to room temperature. The obtained products were centrifuged, washed with deionized water and dried in air at 60 °C for one day.
|
Fig. 1 Schematic diagram of the synthesis of 513MHSH from sea bittern. |
Chemical compositions of the sea bittern
Sea bittern is a solution where sodium ions (Na+) in seawater are partially removed by an ion exchange membrane, resulting in a solution primarily composed of magnesium ions (Mg2+). To determine the components of the sea bittern, Inductively Coupled Plasma Optical Emission Spectroscopy (ICP-OES) analysis was performed. Table 2 shows the ion content results of the sea bittern used in the experiment. The analysis results indicate that the sea bittern contains 3.65 wt% of Mg2+ ions, and it also has a high concentration of other impurities such as Ca2+ and K+ ions. The reason is both Mg2+ ions and impurities like Ca2+ ions from seawater were concentrated when passing through the ion exchange membrane. For use as a Mg precursor, Ca2+ ions must be removed.
Synthesis of 513 MHSH
In Table 1, samples 1-3 were prepared by adding 190 g of H2SO4 to the sea bittern, followed by the addition of MgSO4 and 65-80 ml of 4M NH4OH. Samples 4-6 were prepared by adding 190 g of H2SO4 to the sea bittern and then adding 46-70 ml of 4M NH4OH to a solution of MgSO4 that had been concentrated to approximately 30% through evaporation. Samples 1-6 were stirred for 2 hours and then subjected to hydrothermal synthesis at 180 ℃ for 48 hours in a Teflon vessel. Samples 7-9 were prepared by mixing the Mg(OH)2 solution with H2SO4, followed by stirring and then undergoing hydrothermal synthesis at 180 °C for 24 hours. The precipitate was collected by centrifugation, washed with distilled water, and dried at 60 °C.
Analysis of MgSO4 solution extracted from the sea bittern
A MgSO₄ solution was obtained by adding sulfuric acid to the sea bittern, effectively removing the Ca²⁺ impurities from the Mg precursor. This process utilized the difference in the solubility of MgSO4 and CaSO4. When we added ammonia to the MgSO4 solution from which the precipitated CaSO4 was removed, we obtained Mg(OH)2.
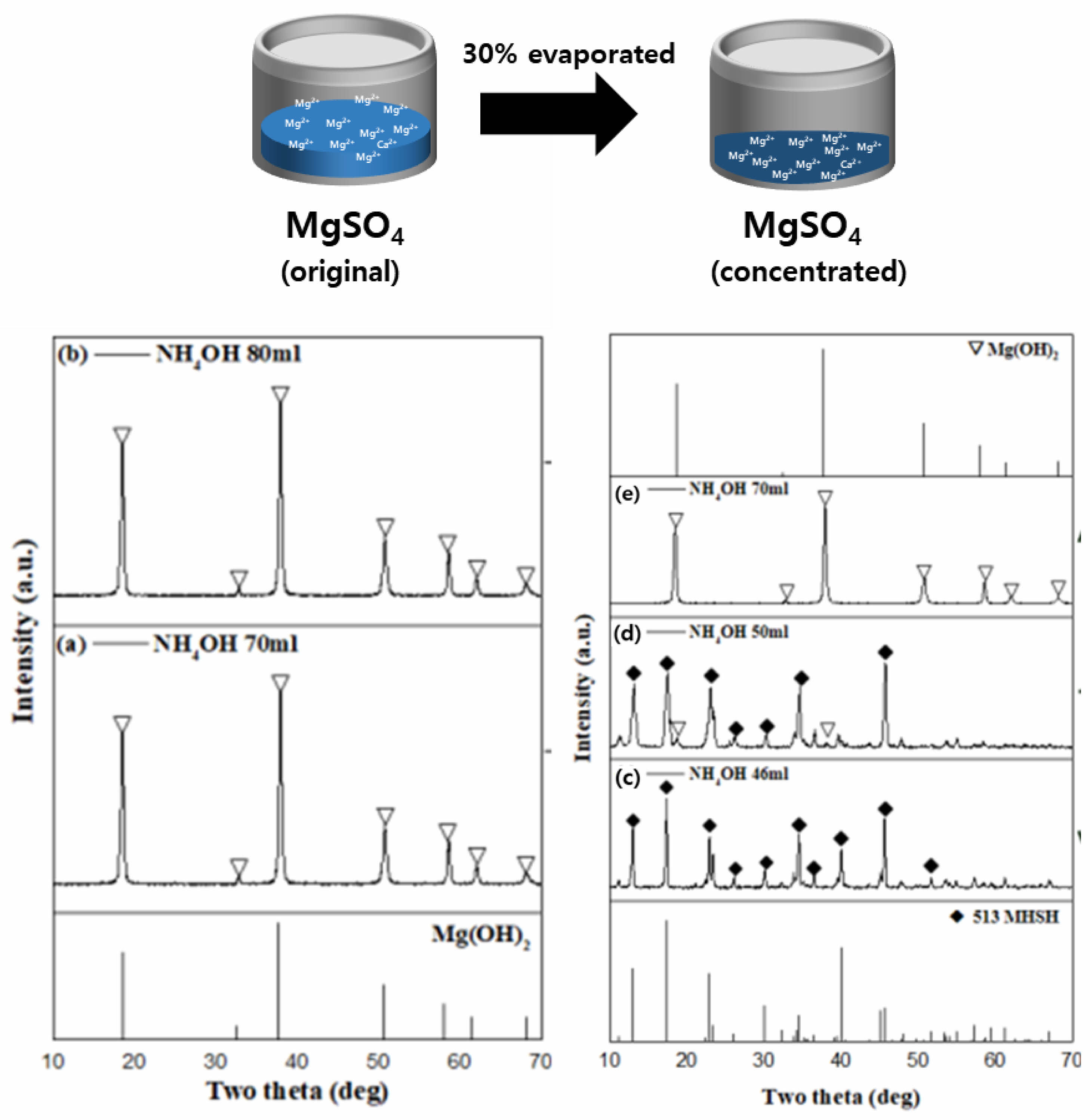
The addition of H2SO4 to sea bittern reduced the Ca2+ ion content by over 90% due to the precipitation of CaSO4 as an insoluble salt from the reaction between the Ca2+ and SO42- ions and the Mg/Ca ratio increased. The removal of Ca²⁺ ions through the reaction with SO₄²⁻ ions enhances the selectivity for Mg²⁺, facilitating the formation of 513 MHSH. Additionally, the addition of sulfuric acid to sea bittern provides SO42− ions, which is an important factor in the formation of 513 MHSH. The ratio of MgSO4/MgO also has a significant effect on the formation of 513 MHSH. The concentration of sea bittern was adjusted based on our previous studies [27]. The MgSO₄ solution derived from the sea bittern was concentrated by approximately 30% through evaporation to increase the Mg²⁺ ion content and facilitate the synthesis of 513 MHSH.
Influence of NH4OH (basic catalyst) on the Formation of 513 MHSH
Figure 2 shows the XRD patterns of the products obtained by varying the amount of NH4OH added to the MgSO4 solution prepared by treating sea bittern with H2SO4. It can be confirmed that when 70 to 80 ml of 4M NH4OH was added to the original sea bittern solution, a single-phase Mg(OH)2 peak was observed (Fig. 2a, 2b). When less than 65 ml of NH4 OH was added, no product formed. To address this result, some of the MgSO4 solutions were evaporated to match the concentration of MgSO4 to the previously published 513 MHSH synthesis conditions [20]. The concentration of the MgSO4 solution from the sea bittern was compared with the previous synthesis data, revealing that NH4OH in a high concentration of MgSO4 significantly affects the shape of the 513 MHSH whiskers. Thus, the MgSO4 concentration from the sea bittern was too low to produce Mg compounds. When using a concentrated MgSO4 solution by evaporating 30% of the original MgSO4 solution, we could obtain the 513 MHSH by adjusting the amount of ammonia.
SEM confirmed these results. Fig. 3 shows the SEM images of the products formed by controlling the amount of NH4OH added to the MgSO4 solution derived from the sea bittern treated with H2SO4. Fig. 3a and 3b show SEM images of aggregated plate-like Mg(OH)2 particles formed from the original sea bittern, while the morphology of products obtained from the concentrated MgSO4 solution varies with the amount of added NH4OH. As the amount of added NH4OH was decreased, the ratio of the plate-like Mg(OH)2 decreased, and the ratio of the needle-like 513 MHSH increased. When 70 ml of 4M NH4OH was added, single-phase Mg(OH)2 was synthesized. The presence of plate-like aggregated materials was confirmed by SEM (Fig. 3e). When 50 mL of 4M NH4 OH was added, Mg(OH)2 and 513 MHSH existed in a mixed state (Fig. 3d). When 46 ml of NH4OH was added, 513 MHSH with a needle-like structure with a diameter of 0.5-1.5 µm and a length of 15-20 µm was observed (Fig. 3c). The pH of the reaction solution significantly affects the dissolution equilibrium of Mg(OH)2 and controls the concentrations of Mg2+ and MgOH+ ions in the solution [20, 21]. When 46, 50, and 70 ml of NH4OH were added, the pH of the solution remained constant at 10. The Mg2+/OH- ratio was controlled without changing the pH by Le Chatelier's principle because a weak base was used. When a strong base solution is used, Mg(OH)2 is generated preferentially, which makes it challenging to obtain high-purity 513 MHSH.
Synthesis of Mg(OH)2 from the sea bittern
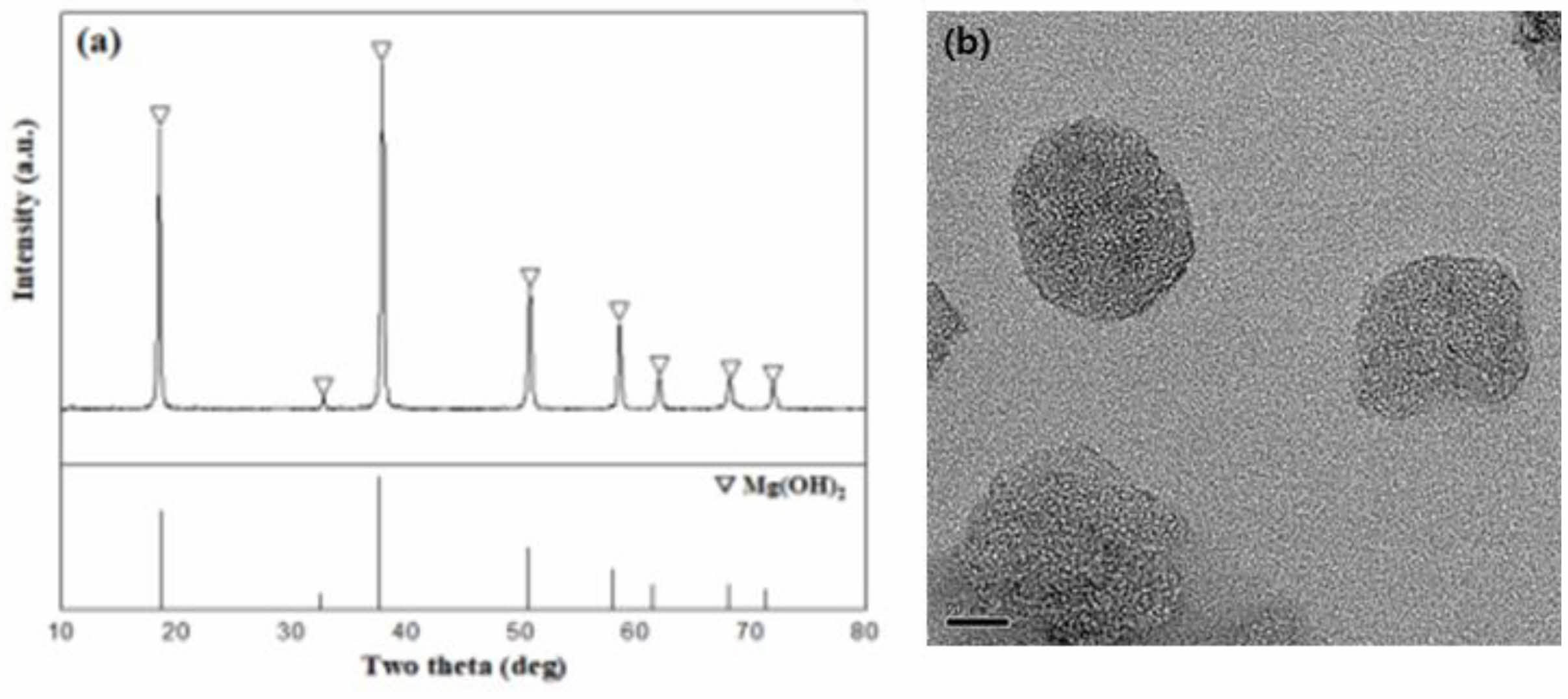
Mg(OH)2 was synthesized by adding ammonia to the MgSO4 from the sea bittern at room temperature. The OH- ions combine with Mg2+ ions in the solution, precipitating as Mg(OH)2. Fig. 4 shows the XRD pattern (Fig. 4a) and TEM image (Fig. 4b) of the Mg(OH)2 from the sea bittern. Adding 2 M NaOH resulted in the formation of Mg(OH)2, confirmed as plate-like particles roughly 50-70 nm in size.
Mg(OH)2 particle growth in a basic condition through hydrothermal treatment
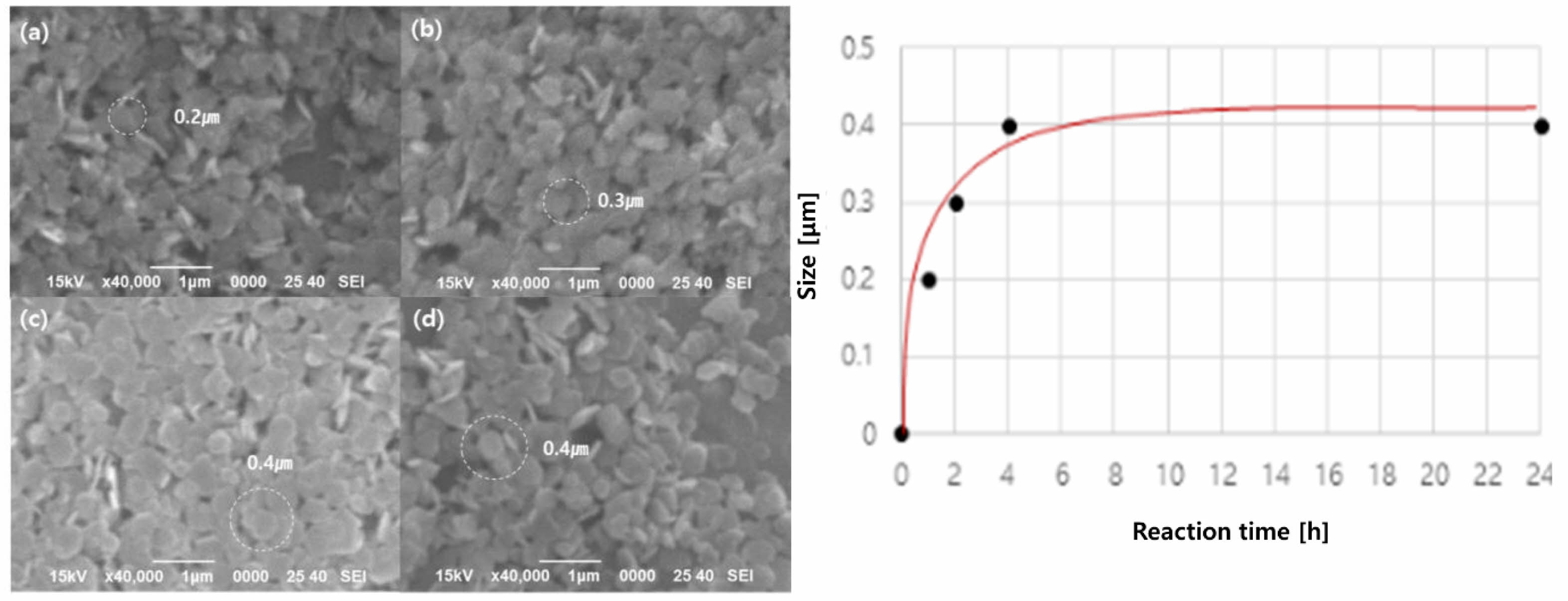
Mg(OH)2 obtained by adding a basic precipitant to sea bittern was dispersed in a 2 M ammonia solution, and the size of the Mg(OH)2 particles was increased through a hydrothermal reaction at 180 ℃. Fig. 5 shows the SEM images and data showing the change in the size of the Mg(OH)2 particles according to the hydrothermal reaction time. It was confirmed that the particle size of Mg(OH)2 (50 nm) increased as the hydrothermal reaction time was extended (Fig. 5a, 5b, 5c). However, the particle size reached approximately 400 nm after 4 hours of hydrothermal treatment and showed no significant growth beyond that point (Fig. 5d). Fig. 5e shows the trend in the Mg(OH)2 particle size change over time. Therefore, it was found that the optimal hydrothermal reaction condition for growing the particles of Mg(OH)2 was a reaction at 180 ℃ for 4 hours.
This result was also observed when magnesium hydroxide was synthesized by excessive addition of NH4OH during 513 MHSH synthesis. This result is because MgOH+ was precipitated around the initial Mg(OH)2 particles and grew as hexagonal plate-like particles as the dissolution and re-precipitation process occurred in the hydrothermal vessels. This result suggests that Mg(OH)2 synthesis and particle growth can be achieved simultaneously [23].
Influence of H2SO4 (acid catalyst) on the formation of 513 MHSH whiskers
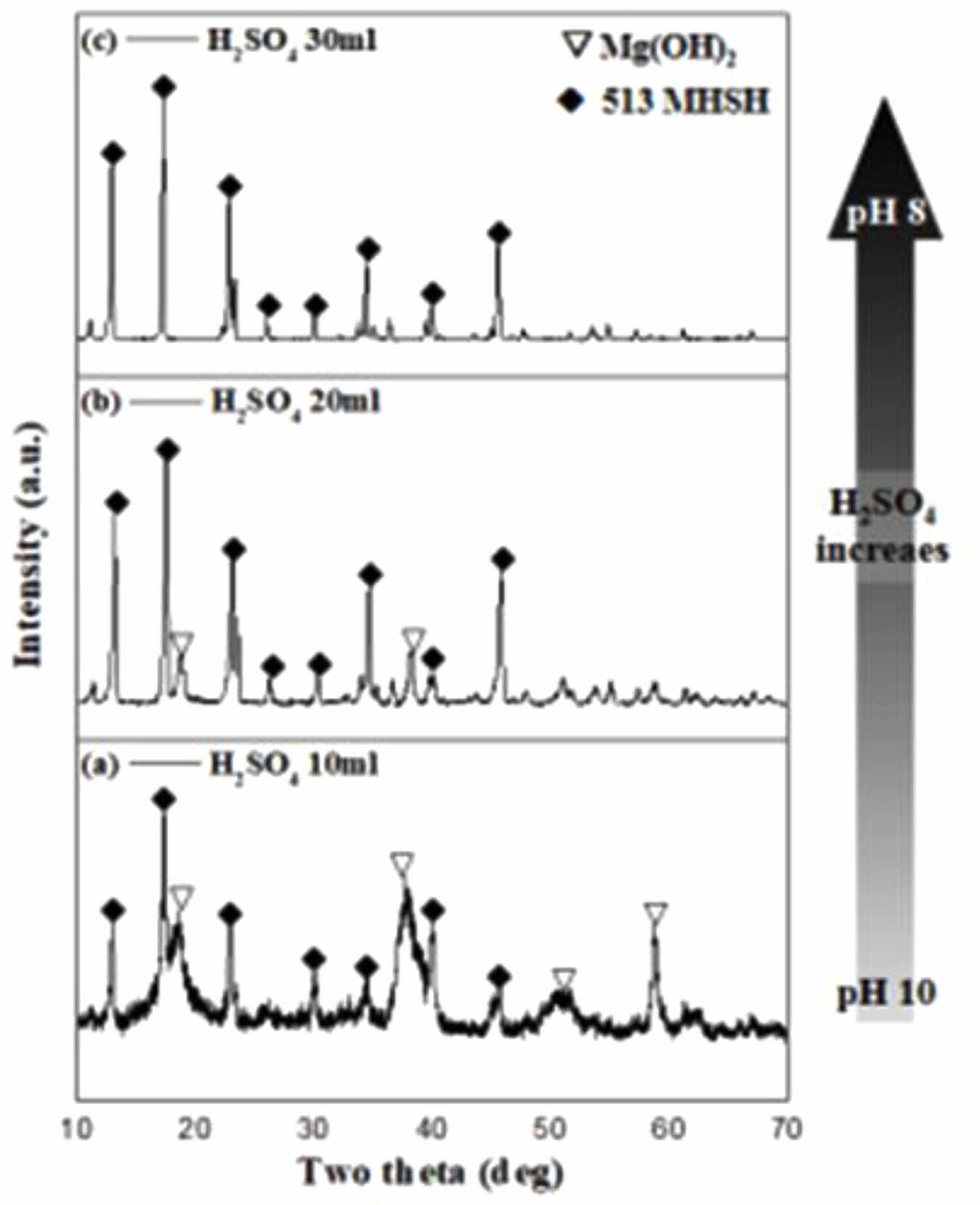
Figure 6 and 7 present the XRD patterns and SEM images of the products obtained using H2SO4 and the Mg(OH)2 derived from the sea bittern. As shown in Fig. 6, an increase in the amount of H2SO4 reacting with the Mg(OH)2 solution led to a decrease in the XRD peak of Mg(OH)2. Specifically, when 30 mL of a 5M H2SO4 solution was added, predominantly needle-like 513 MHSH structures were observed (Fig. 6a). The addition of 10 and 20 mL of the 5M H2SO4 solution resulted in a mixture of Mg(OH)2 and 513 MHSH (Fig. 6b and 6c). The pH decreased within the weak alkaline range from pH 8 to 10 as the H2SO4 concentration was increased, leading to the disappearance of Mg(OH)2 and the formation of pure 513 MHSH. This phenomenon occurs because the solubility of Mg(OH)2 increases as the pH decreases [29]. Consequently, it can be inferred that an increased amount of H2SO4 results in higher concentrations of Mg2+ and MgOH+ due to the dissolution of Mg(OH)2, thereby facilitating the formation of 513 MHSH [18, 19].
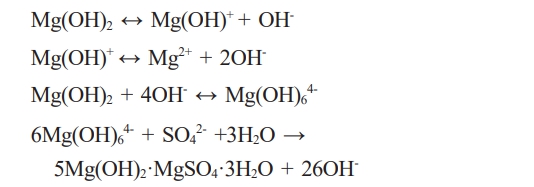
SEM confirmed these results. Fig. 7 shows that the ratio of needle-like 513 MHSH increased when more H2SO4 was added to the Mg(OH)2. In Fig. 7b and 7c, the hexagonal plate-like Mg(OH)2 is dominant, while Fig. 7a shows only the needle-like 513 MHSH.
|
Fig. 2 XRD patterns of the products synthesized at different MgSO4 concentration and ammonia: (a) 70 ml, (b) 80 ml at original MgSO4 [left], (c) 46 ml, (d) 50 ml, (e) 70 ml at evaporated MgSO4 [right]. |

|
Fig. 3 SEM images of the products synthesized at different MgSO4 concentration and ammonia: (a) 70 ml, (b) 80 ml at original MgSO4 [left], (c) 46 ml, (d) 50 ml, (e) 70 ml at evaporated MgSO4 [right]. |
|
Fig. 4 (a) XRD pattern of the products synthesized with sea bittern, (b) TEM image of Mg(OH)2 obtained from sea bittern. |
|
Fig. 5 SEM images of the Mg(OH)2 after hydrothermal treatment in 2 M NaOH 180 ℃ at different reaction time: (a) 1 h, (b) 2 h, (c) 4 h, (d) 24 h, (e) the data of particle size growth according to the hydrothermal reaction time. |
|
Fig. 6 XRD patterns of the products synthesized with 4 M Mg(OH)2 and varying volumes of H2SO4 solution: (a) 30 ml, (b) 20 ml, (c) 10 ml |

|
Fig. 7 SEM images of the products synthesized with 4 M Mg(OH)2 and varying volumes of H2SO4 solution: (a) 30 ml, (b) 20 ml, (c) 10 ml |

This study controlled the shape and size of Mg(OH)2 and 513 MHSH by efficiently extracting MgSO4 from sea bittern and controlling the amount of ammonia. The MgSO4 solution obtained by precipitating Ca2+ as CaSO4 through the addition of sulfuric acid to sea bittern is an important raw material for synthesizing needle-like 513 MHSH. Because the Mg concentration of sea bittern is generally lower than the Mg2+ ion concentration required for synthesis, needle-like 513 MHSH can be synthesized by controlling the Mg content and the amount of ammonia. In addition, ammonia or NaOH, which is a basic precipitant, is added to obtain Mg(OH)2 as hexagonal plate-like particles 50 nm in size, which can be grown to 400 nm through a hydrothermal reaction. In addition, sulfuric acid can be added to the hexagonal plate-like Mg(OH)2 to synthesize the needle-like 513 MHSH through a hydrothermal reaction. In this study, Ca2+ ions present in sea bittern were effectively removed using sulfuric acid. MgSO4 was synthesized and, with the use of a basic catalyst, used to produce both needle-like 513 MHSH and hexagonal plate-like Mg(OH)2. Additionally, sulfuric acid was added to Mg(OH)2 to adjust the Mg(OH)2 to MgSO4 ratio, enabling the formation of the needle-like 513 MHSH. In this study, 513 MHSH was successfully synthesized by controlling the pH through the direct addition of precipitants to the solution without requiring purification or drying of the MgSO4 raw material derived from sea bittern. Furthermore, 513 MHSH can be synthesized by exploiting the reversible conversion between MgSO4 and Mg(OH)2, using basic/acidic catalysts, both of which can be derived from sea bittern. This approach highlights the potential for domestic production of high-value-added compounds from low-cost resources through appropriate technological development.
This work was supported by a grant from “A pilot project for domestic production of magnesium based ceramic raw material” (20016812) funded by the Ministry of Trade, Industry and Energy (MOTIE), Republic of Korea.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
The data used in this study are available upon reasonable request. Interested parties can contact the corresponding author to obtain the data. The data will be shared in a format that is suitable for further analysis, subject to any applicable data use agreements or ethical constraints.
- 1. H. Lu, Y. Hu, L. Yang, J. Xiao, Z.Wang, Z. Chen, and W. Fan, Macromol. Mater. Eng.289[11] (2004) 984-989.
-
- 2. H. Lu, Y. Hu, L. Yang, Z. Wang, Z. Chen, and W. Fan, J. Mater. Sci. 41 (2006) 363-367.
-
- 3. A. Choi, N. Oh, and Y. Kim, J. Korean Ceram. Soc. 59 (2022) 224-228.
-
- 4. Y. Tanga and X. Yuea, J. Ceram. Process. Res. 26[2] (2025), 299-306.
-
- 5. C. Gao, X. Li, L. Feng, Z. Xiang, and D. Zhang, Chem. Eng. J. 150[2-3] (2009) 551-554.
-
- 6. X. Ning, C. Wu, and H. Chen, Materials 15[22] (2022) 8018.
-
- 7. K. Kang and D. Lee, J. Ind. Eng. Chem. 20[4] (2014) 2580-2583.
-
- 8. Y. Tao, G. Shiyang, Z. Lixia, X. Shiuping, and Y. Kaibei, J. Mol. Struct.616[1-3] (2002) 247-252.
-
- 9. E. Kim, Y. Kim, and J. Nam, J. Mater. 4[2] (2018) 149-156.
-
- 10. H. Iwanaga, M. Kawaguchi, and S. Motojima, Jpn. J. Appl. Phys. 32 (1992) 105.
-
- 11. J. Park, S. Kang, and W. Kim, J. Ceram. Process. Res. 10[3] (2009) 364-366.
-
- 12. H. Guo and D. H. Yoon, J. Ceram. Process. Res. 6[4] (2005) 321-325.
- 13. H.M. Lim and J. Yun, J. Ceram. Process. Res. 10[4] (2009) 571-576.
-
- 14. R.S. Plentz, M. Miotto, E.E. Schneider, M.C. Forte, R.S. Mauler, and S.M.B. Nachtigall, J. Appl. Polym. Sci. 101[3] (2006) 1799-1805.
-
- 15. H. Gui, X. Zhang, W. Dong, Q. Wang, J. Gao, Z. Song, J. Lai, Y. Liu, and F. Huang, Polymer 48[9] (2007) 2537-2541.
-
- 16. L. Qiu, R. Xie, P. Ding, and B. Qu, Compos. Struct. 62[3-4] (2003) 391-395.
-
- 17. J. Lv, L. Qiu, and B. Qu, J. Cryst. Growth 267[3-4] (2004) 676-684.
-
- 18. X. Yan, D. Xu, and D. Xue, Acta Mater. 55[17] (2007) 5747-5757.
-
- 19. X. Sun, W. Shi, L. Xiang, and W. Zhu, Nanoscale Res. Lett. 3 (2008) 386.
-
- 20. R. Yu, J.H. Pee, H.T. Kim, K.J. Kim, Y.W. Kim, and Y.J. Kim, Key Eng. Mater. 512 (2012) 91-94.
-
- 21. R. Yu, J.H. Pee, H.T. Kim, K.J. Kim, Y.W. Kim, W. Kim, and Y.J. Kim, J. Korean Powder Metall. Inst. 18[3] (2011) 283-289.
-
- 22. R. Yu, J.H. Pee, H.T. Kim, and Y. Kim, J. Korean Ceram. Soc. 50[3] (2013) 201.
-
- 23. R. Yu and Y. Kim, J. Korean Powder Metall. Inst. 26[3] (2019) 195-200.
-
- 24. M. Park, A. Choi, S. Kim, W. Shim, and Y. Kim, J. Korean Ceram. Soc. 59 (2022) 869-875.
-
- 25. H. Shim, J. Kim, A. Choi, N. Oh, and Y. Kim, J. Korean Powder Metall. Inst. 32[2] (2025) 122-130.
-
- 26. J. Chen, L. Lin, Y. Song, and L. Shao, J. Cryst. Growth 311[8] (2009) 2405-2408.
-
- 27. C. Henrist, J.P. Mathieu, C. Vogels, A. Rulmont, and R. Cloots, J. Cryst. Growth 249[1-2] (2003) 321-330.
-
- 28. Y. Zhang, Z. Yang, D. Guo, H. Geng, and C. Dong, Procedia Environ. Sci. 18 (2013) 84-91.
-
- 29. H. Schott, J. Pharm. Sci. 70[5] (1981) 486-489.
-
 This Article
This Article
-
2025; 26(4): 672-679
Published on Aug 31, 2025
- 10.36410/jcpr.2025.26.4.672
- Received on Jun 3, 2025
- Revised on Jul 6, 2025
- Accepted on Jul 8, 2025
Services
- Abstract
introduction
experimental procedures
results and discussion
conclusion
- Acknowledgements
- Conflict of Interest
- Data availability statement
- References
- Full Text PDF
Shared
Correspondence to
- YooJin Kim
-
Engineering Materials Center, Korea Institute of Ceramic Engineering and Technology, Icheon 17303, Republic of Korea
Tel : +82-31-645-1427 Fax: +82-31-645-1485 - E-mail: yjkim@kicet.re.kr
Clean-Energy Research Institute(CRI), Hanyang University, 222, Wangsimni-ro, Seongdong-gu, Seoul, 04763, Korea
E-mail: jcpr@hanyang.ac.kr