





- Effect of temperature on the electrochemical performance of lithium iron phosphate/carbon cathode materials for lithium-ion batteries
Zhuoer Fu*
School of Engineering and Materials Science, Queen Mary University of London, London, E1 4NS, UK
This article is an open access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
In the development of lithium iron phosphate/carbon (LiFePO4/C), a cathode composite for lithium-ion batteries, essential raw materials including lithium carbonate (Li2CO3), iron phosphate (FePO4), and glucose (C6H12O6) were processed using a high-temperature solid-phase method. This study provided insight into the synthesis process, particularly examining how the heating mode and target reaction temperature critically affected the formation of LiFePO4/C. This study employed Various characterization tool and evaluation systems of X-Ray Diffraction (XRD), Scanning Electron Microscopy (SEM), Thermogravimetric Analysis (TGA) and additional performance evaluations like charge/discharge testing were employed. The findings highlight that the electrochemical properties of lithium iron phosphate/carbon (LiFePO4/C) composites are significantly affected by the target temperature, heating method, carbon content, and degree of graphitization of carbon coating on the material. It was observed that indirect heating methods combined with a target temperature of 750 ℃ optimized the electrochemical performance of materials, enhancing both its discharge capability and electrical capacity. This optimized synthesis approach underscores the intricate balance required in the thermal and compositional parameters to achieve superior battery performance.
Keywords: Lithium iron phosphate/carbon (LiFePO4/C), Heating mode, Target temperature, Electrochemical properties.
As the global population continues to rise, the demand for energy grows correspondingly, exacerbating problems such as environmental pollution and energy shortages. These pressing challenges pose significant threats to the sustainable development of human societies and necessitate urgent attention. Presently, most of the energy supply is sourced from fossil fuels, a non-renewable resource around the world [1-3]. The extensive extraction of fossil fuels, combined with geopolitical instability, increases the risk of an energy crisis. Moreover, the use of fossil fuels leads to significant environmental pollution, contributing to severe issues such as the greenhouse effect, acid rain, and soil and water contamination [4, 5].
Today, energy crises and environmental disasters have a profound impact on human life and development. With growing awareness of the need for energy conservation and environmental protection, the concept of building a low-carbon society has gained global consensus. For the long-term well-being of the planet and its inhabitants, transitioning to clean and renewable energy sources is imperative. Renewable sources such as solar, wind, hydroelectric, biomass, marine, tidal, and geothermal energy can be converted into electricity without the harmful effects associated with fossil fuels [6, 7]. The advancement of energy storage technologies is crucial for mitigating environmental pollution and alleviating the energy crisis [8, 9]. The accelerated development, application, and retirement of energy devices directly influence these critical issues.
In recent years, there has been significant focus on improving the electrochemical performance of lithium-ion battery cathodes, particularly lithium iron phosphate (LiFePO4), by optimizing both particle morphology and surface conductivity [10]. The incorporation of conductive additives, such as carbon black, has been demonstrated to enhance the electronic conductivity of LiFePO4, which in turn improves its rate performance and cycling stability, surface modifications using phosphate coatings, such as Li3PO4 [11], have been explored to stabilize the structure of high-energy cathode materials like Ni-rich LiNi0.8Co0.1Mn0.1O2 [12], thus enhancing their long-term cycling performance highlight the importance of optimizing both the conductivity and structural integrity of cathode materials, offering a promising route for improving the performance of lithium-ion batteries.
Lithium iron phosphate (LiFePO4) is renowned for its longevity, safety, cheap, and non-toxic nature [13, 14]. It plays a crucial role as a cathode material in lithium-ion batteries and has found extensive applications in the electric vehicle sector [15-19]. In the synthesis of LiFePO4 electrode materials for lithium-ion batteries, laboratories primarily utilize solution chemistry methods such as sol-gel [20-23], template [24, 25], spray pyrolysis [26], and hydrothermal processes [27]. These techniques are favored for their ability to precisely customize the dimensions and morphologies of cathode structures, making them effective for creating porous LiFePO4. However, compared to solid-phase reactions, these solution-based methods involve more complex synthesis conditions that are difficult to control and are not easily scalable for industrial application. Consequently, they are not deemed commercially advantageous. In contrast, high-temperature solid-phase synthesis offers a lower production cost and a simpler process, which are more readily adaptable for industrial-scale production [28-30].
However, its poor lithium-ion conductivity and low tap and bulk densities impede the commercial development of high-energy-density LiFePO4 batteries. While numerous researchers have endeavored to enhance the performance of lithium iron phosphate, few have investigated the impact of temperature on its properties.
Temperature significantly affects the particle size, specific surface area, and carbon coating efficiency of materials, thereby influencing the electrochemical performance of composites. It is crucial to emphasize that when the reaction temperature exceeds 800 ℃, the formation of Fe2O3 and Li3Fe(PO4)3 occurs, necessitating that both the calcination temperature and duration be maintained within a reasonable range [31, 32]. Excessively prolonged calcination can lead to particle growth and agglomeration, negatively impacting the performance of lithium iron phosphate (LFP) materials. Managing these parameters is crucial for preserving structural integrity and enhancing the electrochemical properties of LFP, ensuring its suitability for high-performance applications. Ju Guo [33] involved pre-calcining samples in a nitrogen atmosphere at 350 ℃ for 4 h, followed by calcination at 750 ℃ for 8 h. This method addressed issues of poor carbon coating efficiency and high sintering rates commonly encountered in LFP/C synthesis, thereby improving electrochemical performance. The initial specific discharge capacity increased by 3.58% and 9.05% at low (0.5 C) and high (10 C) rates, respectively. Dong[34] utilized ferrous oxalate dihydrate (FeC2O4·2H2O), ammonium hydrogen phosphate ((NH4)2HPO4), and lithium fluoride (LiF) as starting materials to prepare spherical LiFePO4/C using a solid-state method at 650 ℃. The spherical LiFePO4/C demonstrated good reversibility, with an initial discharge capacity of 157 mAh/g at 0.1 C and a capacity retention of nearly 151 mAh/g after 50 cycles, equating to a retention rate of 96.5%. However, variations in heating methods and target temperatures across these studies led to differing outcomes.
This study utilized two heating methods and established three target temperatures to optimize the synthesis of lithium iron phosphate. The findings suggest that samples subjected to indirect heating with a target temperature of 750 ℃ exhibit the best performance, achieving the highest capacity. It is hoped that the methodologies employed in this research offer valuable insights for industrial production.
Materials
Li2CO3, FePO4 and C6H12O6 (anhydrous glucose) was procured from Sinopharm Chemical Reagent Co. Ltd. All chemicals used in the experiments were of analytical grade and were utilized without any additional purification.
Synthesis of LiFePO4/C composites
Li2CO3, FePO4, and C6H12O6 powders were ground and placed in a porcelain boat within the center of a horizontal furnace quartz tube. Argon gas (Ar2) was introduced to displace the existing atmosphere in the quartz tube. Two distinct heating protocols were applied to reach set target temperatures of 700 ℃, 750 ℃, and 800 ℃, maintaining each for 15 h. For direct heating, the temperature was raised from room temperature to 130 ℃, held for 30 min, then increased at 5 ℃/min to the target temperature, held for 15 h, and cooled down to room temperature at 5 ℃/min.
For stepwise heating, the temperature was increased at 5 ℃/min to 130 ℃, held for 30 min, then to 400 ℃, held for another 30 min, and finally to the target temperature at the same rate, maintaining for 15 h. After calcination, the samples were vacuumed and packaged under vacuum. The groups were labeled as LFP1-700, LFP1-750, LFP1-800 for direct heating and LFP2-700, LFP2-750, LFP2-800 for stepwise heating.
A portion of the samples was reserved for characterization (SEM, TGA, N2 adsorption-desorption) and the initial grinding process was repeated to prepare samples for cells testing.
Characterization and electrochemical test
The experiment employed the Rigaku D/Max-2500V/PC X-ray diffractometer (XRD) with a Cu Kα radiation source. XRD technology is essential for probing the crystalline structure within materials, enabling the analysis of phase transformations. Data acquisition during the experimental procedure utilized a Philip XL 30 scanning electron microscope, which recorded the surface features, morphology, and cross-sectional structure of the LiFePO4/C composite material. Thermal behavior was analyzed using a Shimadzu Corporation TA-50 thermal analyzer in a nitrogen atmosphere, with a sample mass of approximately 10 mg. The nitrogen flow was maintained at 25 mL/min, with a heating rate of 10 ℃/min up to a maximum temperature of 850 ℃, placing the LiFePO4/C samples in a quartz crucible. A smaller sample size of 6-8 mg was preferred for each measurement to minimize adverse effects from thermal diffusion. Structural analysis of carbonaceous intermediates was conducted using the MKI-2000 type confocal Raman microscope from Renishaw plc Corporation, utilizing a helium-neon laser with a wavelength of 632.8 nm and a power setting of 30 mW, at a 500x magnification. Samples were dried in a vacuum oven at 120 ℃ for 12 h.
Electrochemical testing of the LiFePO4/C samples was conducted using CR2025 coin cells. The electrode material was prepared by mixing active material powder, carbon black, and polyvinylidene fluoride (PVDF) in a ratio of 90:5:5 dispersed in N-methyl-2-pyrrolidone. The mixture was coated onto an Al foil substrate. After drying the coated foil at 80 ℃ under vacuum for 12 h, it was cut into discs of 1.5 cm2, which were then pressed under 10 MPa to form the working electrodes. The final cathode contained approximately 2.3 mg of active material, ensuring uniform thickness across the electrode. Assembly of the cells was performed in an argon-filled glove box, using a Celgard 2400 separator and an electrolyte composed of equal volumes of ethylene carbonate, diethyl carbonate, and dimethyl carbonate, with 1.0 M LiPF6. The assembled cells were left to rest for over 24 h before performance testing at 25 ℃ using the LAND system, with the charge and discharge cycles set between 2.5 V and 4.2 V.
Structure and Morphology of LiFePO4/C
Research has demonstrated that the calcination temperature is critical in synthesizing LiFePO4/C cathode materials for lithium-ion batteries. It is widely accepted that the ideal calcination temperatures are either 700 ℃ or 750 ℃. At these temperatures, LiFePO4 crystals are effectively stabilized and their growth into larger particles is inhibited [35-38]. In this study, to determine the most effective calcination temperature and heating method for LiFePO4/C, we set three target temperatures, 700 ℃, 750 ℃ and 800 ℃, and two types of heating methods, direct and indirect, respectively.
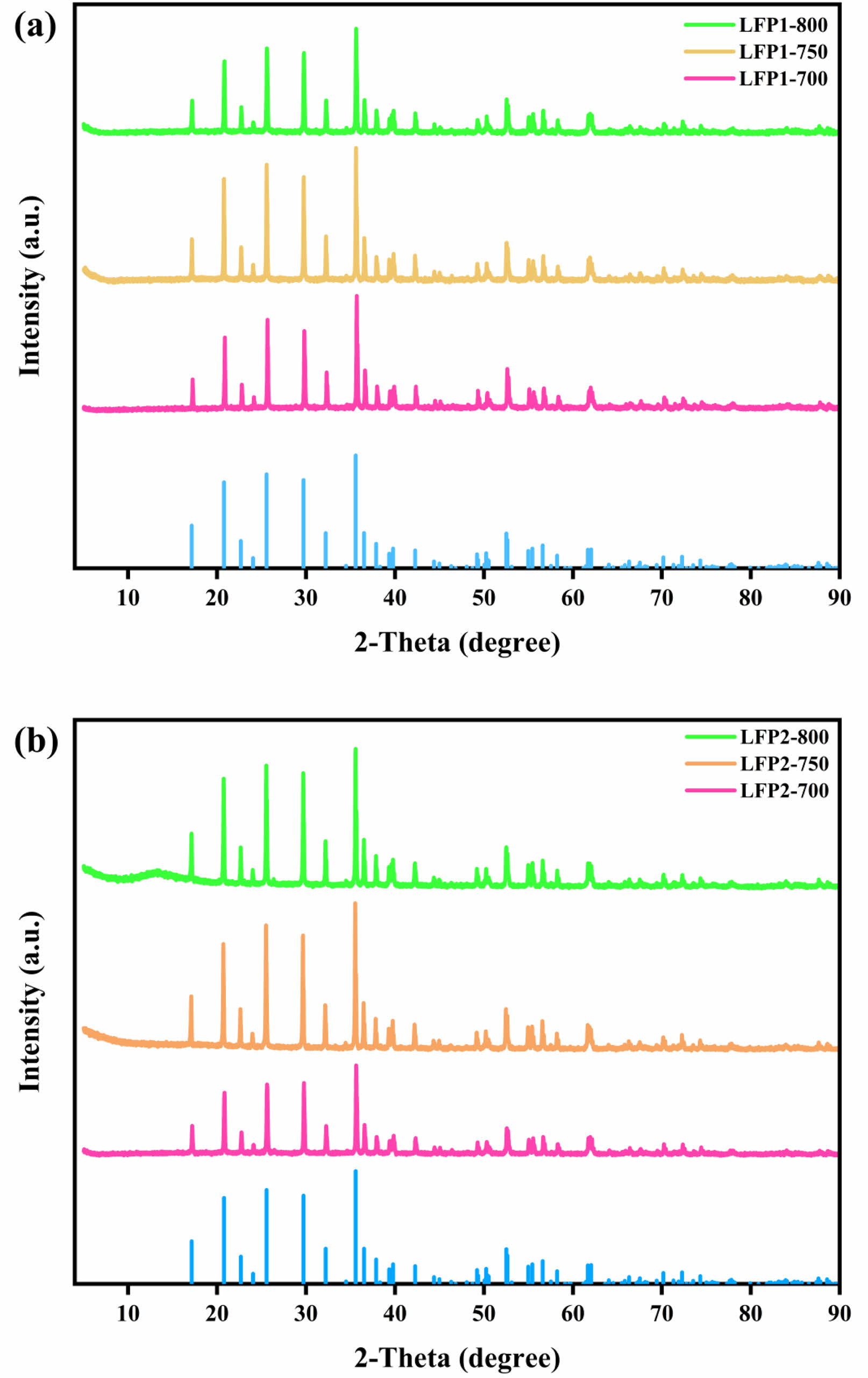
Fig. 1 presents XRD patterns of LiFePO4/C powders prepared via high-temperature solid-state synthesis. The XRD patterns of the two groups of LiFePO4/C composite materials in Fig. 1a and Fig. 1b both show a highly crystalline orthorhombic olivine structure, belonging to the Pnma space group. This configuration aligns with the standard described in JCPDS no. 83-2092. The samples display distinct peaks, indicating good crystallinity, with no impurities detected. Among the LFP1-x samples (x stands for 700 ℃, 750 ℃, and 800 ℃.), LFP1-750 exhibits the highest degree of crystallinity, characterized by sharp and intense peaks. The absence of additional peaks suggests minimal presence of secondary phases, further confirming the high purity of the synthesized materials. Fig. 1b shows that in the LFP2-x samples, the XRD patterns reveal higher crystallinity compared to the LFP1-x samples, with more defined and prominent peaks. This improvement indicates a more ordered crystal structure, which may contribute to the superior electrochemical performance of the LFP2-x samples. Notably, the LFP2-750 sample stands out with the highest peak intensity and clarity, indicating optimal synthesis conditions. Additionally, the XRD patterns show a slight shift of peak positions towards higher angles, suggesting a contraction of the unit cell volume due to more favorable synthesis conditions, leading to enhanced crystallinity and phase purity. Overall, Fig. 1 demonstrates the feasibility of synthesizing LiFePO4/C composites under appropriate carbothermal reduction conditions.
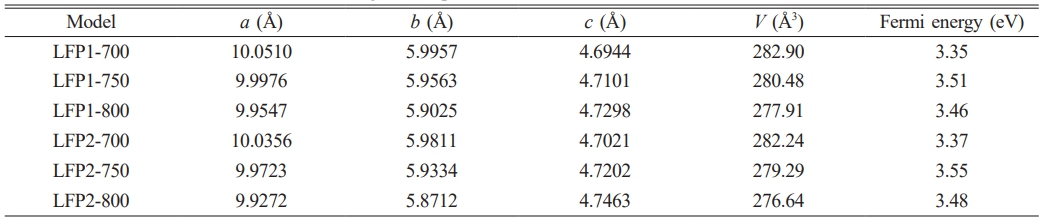
Table 1 presents the optimized lattice parameters and Fermi energy of LiFePO4/C. As shown in Table 1, with increasing temperature, parameters a and b decrease significantly, while lattice parameter c increases slightly. These changes lead to a reduction in the unit cell volume, which may enhance the diffusion of Li+ within the bulk phase [39, 40]. The decrease in parameter a and the increase in parameter c both contribute to reducing the diffusion energy of Li+.
Additionally, Table 1 shows that within the LFP1 series, as the temperature rises to 750 ℃, the Fermi energy increases from 3.35 eV to 3.51 eV. This suggests that moderately increasing the temperature enhances the carbon coating of the LiFePO4/C composite, improving electrical conductivity, thus enhancing rate performance, and inhibiting the growth of LiFePO4 grains, leading to higher energy density [41]. However, with further temperature increases, the Fermi energy decreases, likely due to excessive growth of LiFePO4 particles at high calcination temperatures (e.g., 800 ℃), forming larger particles that hinder Li+ insertion/extraction from the electrode.
Fig. 2 displays SEM images of LiFePO4/C synthesized via the high-temperature solid-state method at 700 ℃, 750 ℃, and 800 ℃. Fig. 2a~Fig. 2f show that the sample synthesized at the target temperature of 750 ℃ exhibits the most uniform particle size with significantly fewer agglomerations. Fig. 2a and Fig. 2d show that the sample synthesized at 700 ℃ shows more uneven particle sizes with fewer agglomerations. Fig. 2c and Fig. 2f show that the sample synthesized at 800 ℃, while having a relatively uniform particle size, displays a higher degree of agglomeration. These results suggest that the high-temperature solid-state method is suitable for producing submicron-sized (approximately 350 nm) LiFePO4/C powders. Furthermore, the particle size and porosity can be effectively controlled by adjusting the target temperature and heating method.
The LiFePO4/C samples underwent detailed elemental analysis using EDX. Fig. 3 shows that the sample synthesized at 750 ℃ does not exhibit significant agglomeration or phase separation. However, compared to Fig. 3a, the sample in Fig. 3b has more uniform particle sizes and significantly fewer agglomerations. This indicates that during the synthesis of LiFePO4/C composites, appropriate heat treatment can lead to more complete carbonization, better crystallinity, and a more intact structure.
The EDX elemental analysis of the two sets of LiFePO4/C synthesized at 750 ℃ in Fig. 3 shows no detectable impurities, indicating that the elements C, Fe, P, and O are uniformly distributed in the sample, resulting in the best performance. Uniform elemental distribution is crucial for the performance of battery materials. In LiFePO₄ cathode materials, the uniform distribution of Fe and P ensures the stability and integrity of the crystal structure, while the even distribution of O guarantees the chemical integrity of materials and electrochemical performance. The homogeneous distribution of the carbon coating significantly enhances the conductivity of materials, ensuring efficient electron transport during the charge and discharge processes [42, 43]. In this study, the optimized synthesis conditions led to the LFP2-750 sample demonstrating excellent electrochemical performance. This sample exhibited a specific capacity exceeding 150 mAh/g and maintained a high-capacity retention rate after numerous charge-discharge cycles.
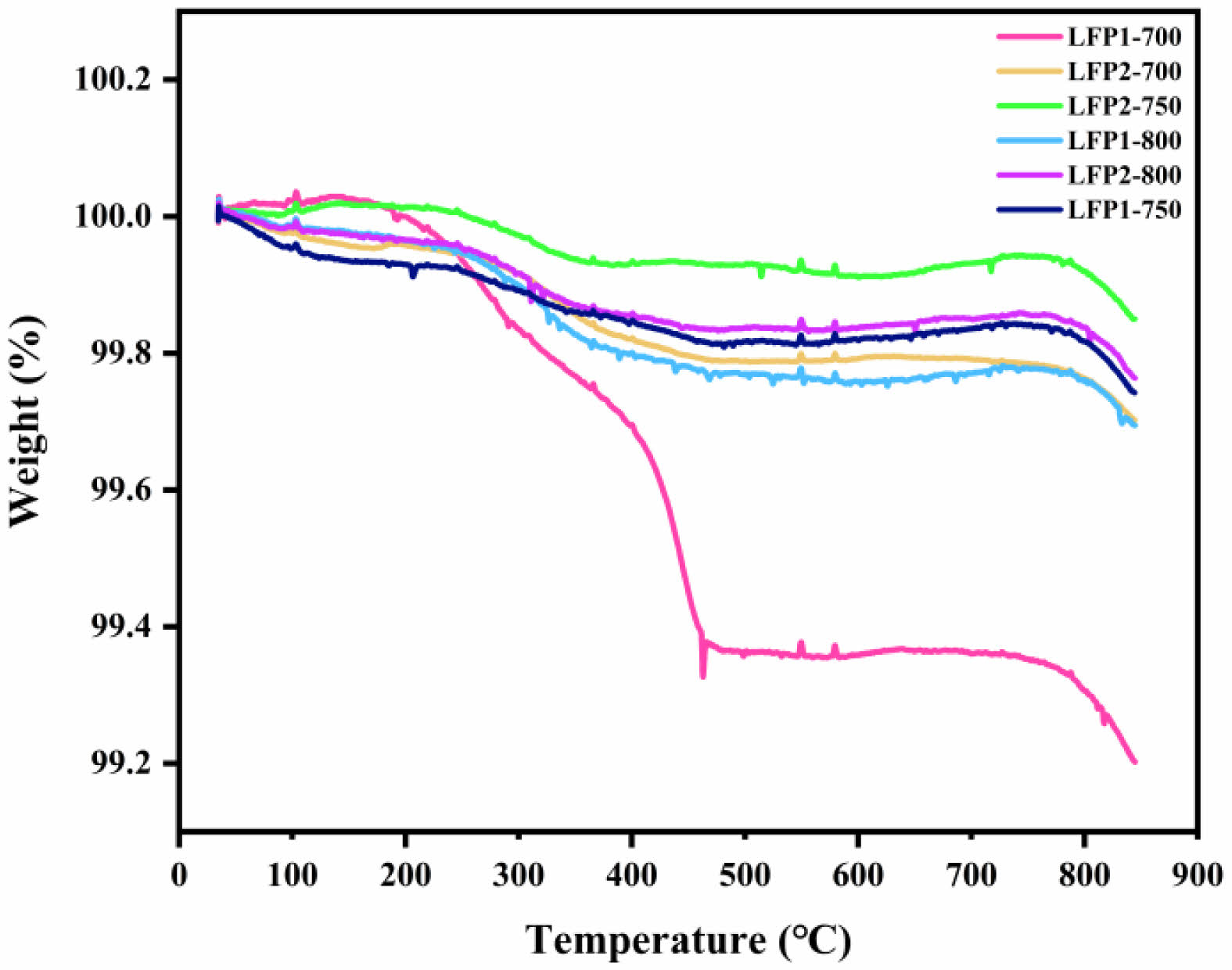
Fig. 4 presents the TGA curve of LiFePO4/C. The LFP1-x and LFP2-x samples exhibit distinct thermal behaviors when heated to 800 ℃. The LFP1-700 sample shows significant weight loss between 300 ℃ and 400 ℃, indicating thermal decomposition of its components or impurities. This rapid weight loss differentiates it from other samples. After exceeding 800 ℃, the sample continues to lose weight, which may indicate a transformation or collapse of the lithium iron phosphate lattice. This structural change could result in poorer electrochemical performance. In contrast, LFP1-750 and LFP1-800 samples exhibit more gradual weight loss over the temperature range, suggesting better thermal stability compared to LFP1-700. Overall, the LFP2-x samples demonstrate superior thermal stability compared to the LFP1-x series. All LFP2-x samples show minimal weight loss, indicating better retention of material integrity at high temperatures. Notably, LFP2-750 stands out as the best performer among the LFP2-x samples, maintaining its weight up to higher temperatures before experiencing a slight decline. This suggests that LFP2-750 has the most optimal synthesis conditions and performance.
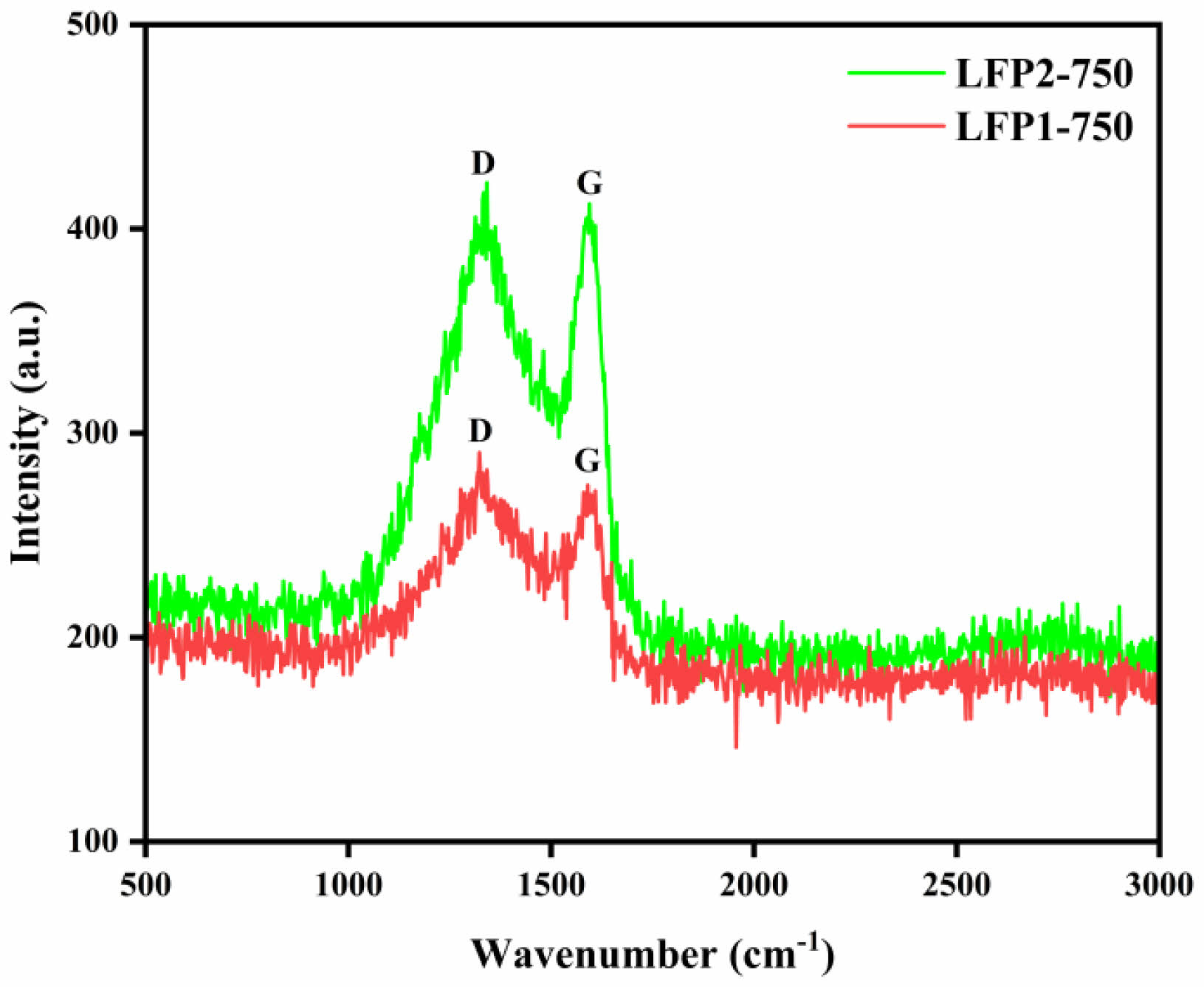
Figure 5 presents the Raman spectra of the LiFePO4/C composites. The peaks at 1323 cm−1 and 1341 cm−1 correspond to the disordered carbon (D band) in LFP1-750 and LFP2-750, respectively. The peaks at 1590 cm−1 and 1594 cm−1 correspond to the graphitized carbon (G band) in LFP1-750 and LFP2-750, respectively. In Raman spectroscopy, the intensity ratio of the D band to the G band (ID/IG) indicates the degree of graphitization; a lower ID/IG ratio signifies a higher degree of graphitization. In this study, the ID/IG ratio for LFP1-750 is 0.880, while for LFP2-750, it is 0.862. This suggests that appropriate heat treatment during the preparation of LiFePO4/C composites leads to more complete carbonization, resulting in less disordered graphitized carbon, which is beneficial for enhancing conductivity.
Electrochemical Performance
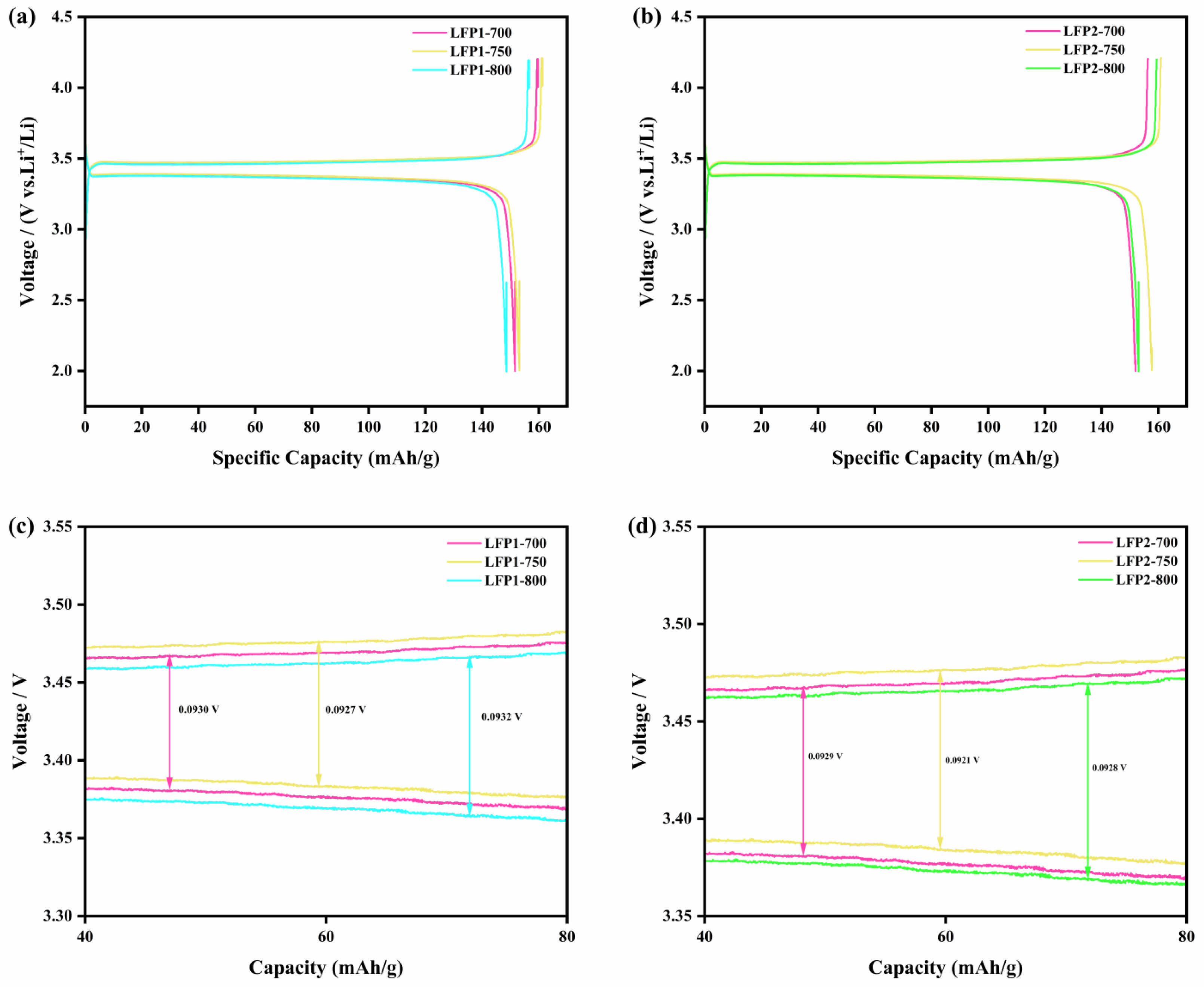
Fig. 6 illustrates the charge-discharge curves and the voltage platform potential differences of LFP1-750 and LFP2-750. Fig. 6a and Fig. 6b depict the charge-discharge profiles for LFP1-750 and LFP2-750 at a current rate of 0.1 C (1 C = 170 mAh/g) within a voltage range of 2.5 V to 4.2 V. The voltage plateau around 3.4 V indicates the stable electrochemical performance of the LiFePO4 composites. Notably, LFP2-750 exhibits an initial discharge capacity of 161.8 mAh/g, surpassing the specific capacity of LFP1-750, which is 160.7 mAh/g. Comparatively, the specific capacity of LFP1-750 is slightly higher than that of LFP1-700 and LFP1-800. A similar trend is observed among LFP2-700, LFP2-750, and LFP2-800. Within the LFP2-x samples synthesized via indirect heating, LFP2-750 demonstrates the highest specific capacity, indicating optimal electrochemical activity at a synthesis temperature of 750 ℃. This suggests that the synthesis method and specific temperature enhance the crystalline structure and conductivity of the material, improving lithium-ion diffusion and electronic conductivity. Fig. 6c and Fig. 6d show the potential differences between the charge and discharge voltage platforms of LFP1-750 and LFP2-750. It is found that the appropriate target temperature and heating method effectively reduce the potential difference, with LFP1-750 at 92.7 mV and LFP2-750 at 92.1 mV. At the same target temperature, the potential difference of indirectly heated samples is marginally lower than that of directly heated samples, indicating that the LiFePO4/C composites produced via suitable methods exhibit faster reaction kinetics.
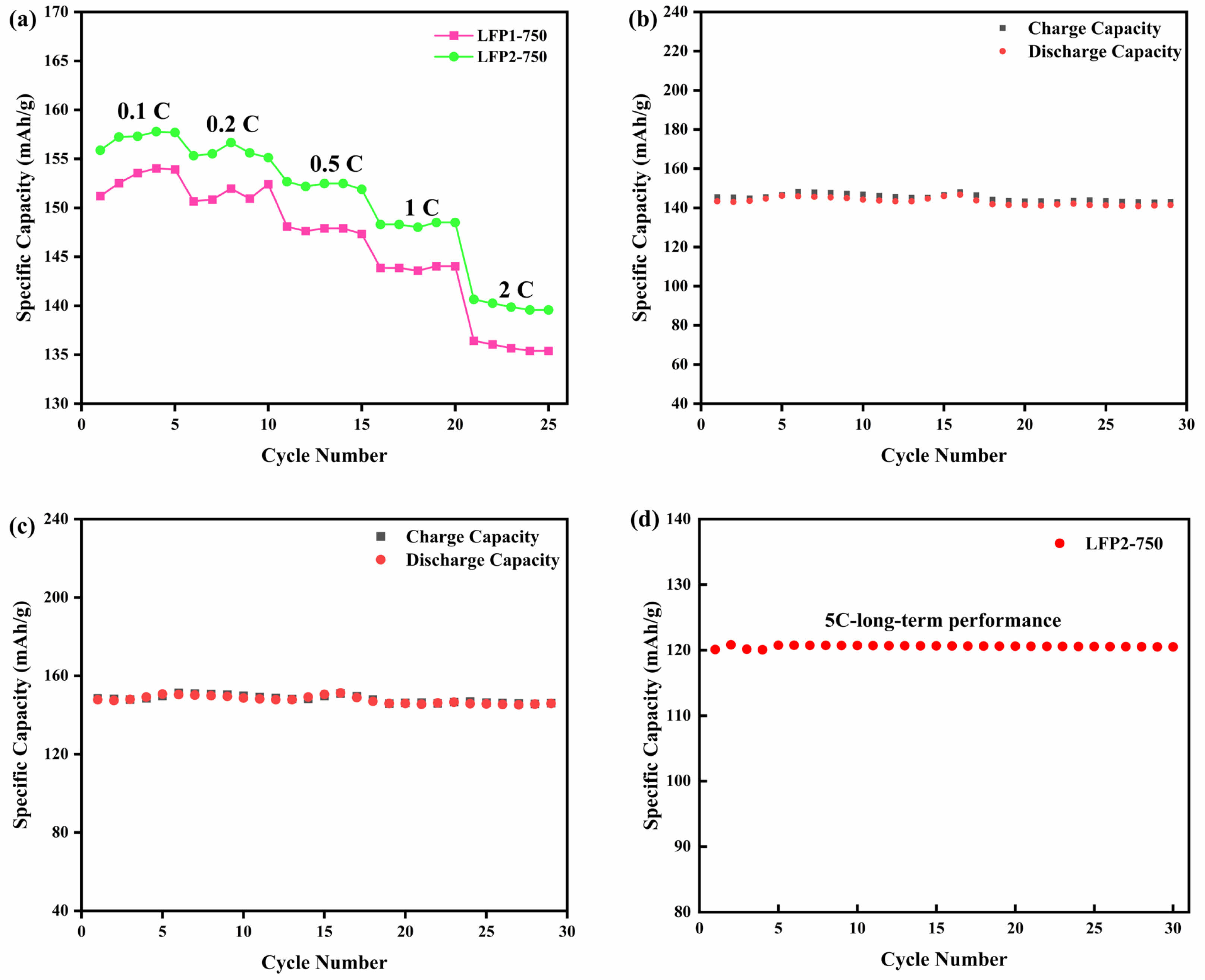
Fig. 7 illustrates the rate performance and cycling stability of LiFePO4/C materials under different current densities. Fig. 7a displays the rate performance of LFP1-750 and LFP2-750 at various rates. The data indicates that as the current density increases from 0.1 C to 2 C, the specific capacities of both materials decline. However, the specific capacity of LFP2-750 remains higher than that of LFP1-750 across all current densities, with a pronounced advantage at higher rates. This suggests that LFP2-750 exhibits superior electrochemical performance and better capacity retention under high-rate conditions. Fig. 7b and Fig. 7c present the cycling performance of LFP1-750 and LFP2-750 at a current density of 1 C. In Fig. 7b, the charge and discharge capacities of LFP1-750 exhibit slight fluctuations with increasing cycle numbers but remain relatively stable overall. In contrast, Fig. 7c shows that LFP2-750 demonstrates higher charge-discharge capacities and less capacity fading under the same conditions, indicating greater stability and cycling performance. Fig. 7d illustrates the long-term cycling stability of LFP2-750 at a challenging 5 C rate. Even under these stringent conditions, the material sustains a stable capacity with minimal fading over numerous cycles. This behavior highlights how optimized synthesis protocol, particularly the stepwise heating approach—confers robust structural integrity and efficient ion/electron transport pathways. The ability to retain a high capacity at 5 C is consistent with the superior performance observed at lower rates, further confirming the synergistic benefits of high crystallinity, uniform particle size, and effective carbon coating. Consequently, LFP2-750 holds considerable promise for applications demanding both high power and prolonged cycle life.
The enhanced electrochemical activity, electronic and ionic conductivity, superior cycling stability and rate performance of LFP2-750 are attributed to its optimized synthesis method and temperature. Notably, the capacity of LFP2-750 retention at high rates significantly outperforms that of LFP1-750, highlighting the substantial advantages of the indirect heating method in enhancing material performance.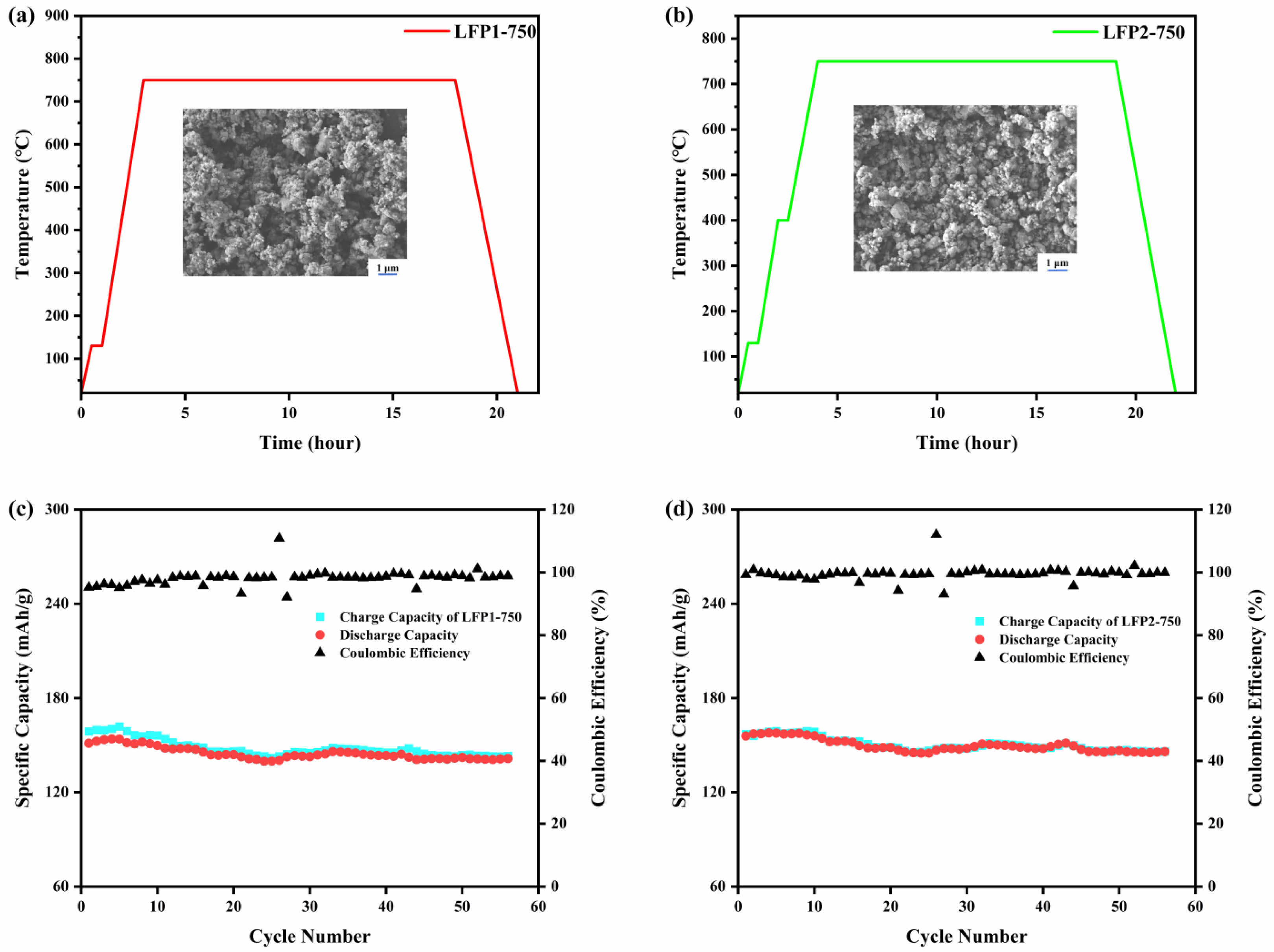
Scheme 1. Heating curves and SEM images of (a) LFP1-750; (b) LFP2-750; charge-discharge rate graphs of (c) LFP1-750; (d)
LFP2-750.
Scheme 1 presents the rate performance, heating curves, and SEM images of LFP1-750 and LFP2-750, offering a detailed comparison of these materials electrochemical performance under different current densities, their heating processes, and microstructures. The high efficiency and stability can be attributed to appropriate particle size and structural optimization, which contribute to the improved electrochemical performance of the material. Scheme 1a indicates the heating profile of LFP1-750, employing a direct heating method where the temperature is maintained at 750 ℃ for 15 h. Conversely, Scheme 1b indicates a different thermal approach for LFP2-750, involving an indirect heating process. This method initially raises the temperature to 400 ℃, holds it for 0.5 h, and then increases it to 750 ℃, maintaining this peak for an additional 15 h. Comparative analysis of scanning electron microscopy images reveals that LFP2-750 exhibits a more uniform and finer particulate structure than LFP1-750.
Scheme 1c indicates that LFP1-750 maintains relatively stable capacity across various rates (0.1 C to 2 C) with a Coulombic efficiency of 98.14%. Similarly, LFP2-750 retains high capacity and efficiency at different cycling rates, achieving a Coulombic efficiency of 99.50%, demonstrating superior performance. Scheme 1d illustrates the performance of LFP2-750 at the same current density, showing superior rate performance with minimal capacity fade. This enhanced performance underscores the advantages of the indirect heating method, which likely improves crystal structure, phase purity, and electronic and ionic conductivity [44].
|
Fig. 1 XRD patterns of each group of LFP/C. |

|
Fig. 2 SEM images of LFP/C samples. (a), (b), and (c) denote LFP1-x, and (d), (e), and (f) denote LFP2-x. x stands for 700 ℃, 750 ℃, and 800 ℃. |

|
Fig. 3 Mapping images of (a) LFP1-750; (b) LFP2-750. |
|
Fig. 4 TGA image of LiFePO4/C. |
|
Fig. 5 Raman spectrum of LiFePO4/C composite calcinated at 750 ℃. |
|
Fig. 6 Typical charge–discharge curves of (a) LFP1-x samples; (b) LFP2-x samples at 0.1 C; the gap between the charge and discharge plateaus of (c) LFP1-x samples; (d) LFP2-x samples at 0.1 C. |
|
Fig. 7 (a) Rate performance graphs of LFP1-750 and LFP2-750 at various rates; Cycling performance of (b) LFP1-750; (c) LFP2- 750 at 1 C; (d) The long-term performance of LFP2-750 at 5 C. |
LiFePO4/C composites were synthesized using the high-temperature solid-state method, and their electrochemical performance as cathode materials for lithium-ion batteries was investigated, focusing particularly on the effects of temperature and heating methods on their electrochemical properties. The results indicate that both temperature and heating method significantly influence the discharge capacity and rate performance of LiFePO4/C composites. The optimal synthesis conditions were identified as a target temperature of 750 ℃ and the use of indirect heating.
Appropriate heat treatment during the synthesis process ensures more complete carbonization, improved crystallinity, and a more intact structure. These findings underscore the significant potential of LiFePO4/C composites as a novel cathode material for lithium-ion batteries.
No ethical approval was required as it did not involve the collection or analysis of data involving human or animal subjects.
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
The authors declared no potential conflicts of interest with respect to the research, author-ship, and publication of this article.
The authors received no financial support for the research.
- 1. C. Kuzemko, M. Blondeel, C. Dupont, and M.C. Brisbois, Energy Res. Soc. Sci. 93 (2022) 102842.
-
- 2. N.Z. Muradov and T.N. Veziroğlu, Int. J. Hydrog. Energy 33[23] (2008) 6804-6839.
-
- 3. L. Schramm, Eur. Political Sci. Rev. 16[1] (2024) 56-71.
-
- 4. A.B. Awan and Z.A. Khan, Renew. Sustain. Energy Rev. 33 (2014) 236-253.
-
- 5. X. Li, H. Wang, Q. Zhou, T. Qi, G. Liu, and Z. Peng, Waste Manag. 87 (2019) 798-804.
-
- 6. S. Maddukuri, D. Malka, M.S. Chae, Y. Elias, S. Luski, and D. Aurbach, O Electrochim. Acta 354 (2020) 136771.
-
- 7. A. Rahman, O. Farrok, and M.M. Haque, Renew. Sustain. Energy Rev. 161 (2022) 112279.
-
- 8. W. Wang and Y. Wu, Resour. Conserv. Recycl. 127 (2017) 233-243.
-
- 9. J. Xiang, Y. Wei, Y. Zhong, Y. Yang, H. Cheng, L. Yuan, H. Xu, and Y. Huang, Adv. Mater. 34[52] (2022) 2200912.
-
- 10. U.J . Jeon, T.-Y. Shim, I.-H. Im, K.-N. Kim, and S.-H. Lee, J. Ceram. Process. Res. 25[6] (2024) 937-940.
-
- 11. Y.N. Luo, Y.T. Tsai, and K.H. Lin, J. Ceram. Process. Res. 17[10] (2016) 1082-1087.
-
- 12. D.-Y. Hwang and S.-H. Lee, J. Ceram. Process. Res. 22[4] (2021) 421-424.
-
- 13. C.-Y. Huang, T.-R. Kuo, S. Yougbaré, and L.-Y. Lin, J. Colloid Interface Sci. 607 (2022) 1457-1465.
-
- 14. X. Lai, Y. Huang, H. Gu, C. Deng, X. Han, X. Feng, and Y. Zheng, Energy Storage Mater. 40 (2021) 96-123.
-
- 15. N. Alias and A.A. Mohamad, J. Power Sources. 274 (2015) 237-251.
-
- 16. M. Armand and J.-M. Tarascon, Nature 451 (2008) 652-657.
-
- 17. S.-Y. Chung, J.T. Bloking, Y.-M. Chiang, Nat. Mater. 1 (2002) 123-128.
-
- 18. S. Huang, J. Ren, R. Liu, Y. Bai, X. Li, Y. Huang, M. Yue, X. He, and G. Yuan, ACS Sustain. Chem. Eng. 6[10] (2018) 12650-12657.
-
- 19. A.K. Padhi, K.S. Nanjundaswamy, J.B. Goodenough, J. Electrochem. Soc. 144[4] (1997) 1188.
-
- 20. R. Dominko, M. Bele, J.-M. Goupil, M. Gaberscek, D. Hanzel, I. Arcon, and J. Jamnik, Chem. Mater. 19[12] (2007) 2960-2969.
-
- 21. S.W. Oh, S.-T. Myung, H.J. Bang, C.S. Yoon, K. Amine, and Y.-K. Sun, Electrochem. Solid-State Lett. 12[9] (2009) A181.
-
- 22. F. Yu, J.-J. Zhang, Y.-F. Yang, and G.-Z. Song, J. Mater. Chem. 19[48] (2009) 9121-9125.
-
- 23. Y. Zhou, J. Wang, Y. Hu, R. O’Hayre, and Z. Shao, Chem. Commun. 46[38] (2010) 7151-7153.
-
- 24. C.M. Doherty, R.A. Caruso, B.M. Smarsly, and C.J. Drummond, Chem. Mater. 21[13] (2009) 2895-2903.
-
- 25. N.N. Sinha, C. Shivakumara, and N. Munichandraiah, ACS Appl. Mater. Interfaces 2[7] (2010) 2031-2038.
-
- 26. J. Liu, T.E. Conry, X. Song, M.M. Doeff, and T.J. Richardson, Energy Environ. Sci. 4[3] (2011) 885-888.
-
- 27. J. Qian, M. Zhou, Y. Cao, X. Ai, and H. Yang, J. Phys. Chem. C 114[8] (2010) 3477-3482.
-
- 28. G.T.-K. Fey, Y.G. Chen, and H.-M. Kao, J. Power Sources 189[1] (2009) 169-178.
-
- 29. G.T.-K. Fey and T.-L. Lu, J. Power Sources 178[2] (2008) 807-814.
-
- 30. B.Q. Zhu, X.H. Li, Z.X. Wang, and H.J. Guo, Physics, Mater. Chem. Phys. 98[2-3] (2006) 373-376.
-
- 31. D. Jugović and D. Uskoković, J. Power Sources 190[2] (2009) 538-544.
-
- 32. M. Takahashi, S.-I. Tobishima, K. Takei, and Y. Sakurai, Solid State Ionics 148[3-4] (2002) 283-289.
-
- 33. J. Guo, C. Liang, J. Cao, and S. Jia, Int. J. Hydrog. Energy 45[58] (2020) 33016-33027.
-
- 34. Y. Dong, Y. Zhao, Y. Chen, Z. He, and Q. Kuang, Mater. Chem. Phys. 115[1] (2009) 245-250.
-
- 35. S.Y. Chung, S.Y. Choi, T. Yamamoto, and Y. Ikuhara, Angew. Chem. Int. Ed. 48[3] (2009) 543-546.
-
- 36. N. Meethong, Y.H. Kao, S.A. Speakman, and Y.M. Chiang, Adv. Funct. Mater. 19[7] (2009) 1060-1070.
-
- 37. X.-L. Wu, L.-Y. Jiang, F.-F. Cao, Y.-G. Guo, and L.-J. Wan, Adv. Mater. 21[25-26] (2009) 2710-2714.
-
- 38. T.-H. Cho and H.-T. Chung, J. Power Sources 133[2] (2004) 272-276.
-
- 39. X. Yin, K. Huang, S. Liu, H. Wang, and H. Wang, J. Power Sources 195[13] (2010) 4308-4312.
-
- 40. D. Zhang, P. Zhang, J. Yi, Q. Yuan, J. Jiang, Q. Xu, Z. Luo, and X. Ren, J. Alloys Compd. 509[4] (2011) 1206-1210.
-
- 41. K.-F. Hsu, S.-Y. Tsay, and B.-J. Hwang, J. Mater. Chem. 14[17] (2004) 2690-2695.
-
- 42. P. Benedek, N. Wenzler, M. Yarema, and V.C. Wood, RSC Adv. 7[29] (2017) 17763-17767.
-
- 43. I. Stenina, D. Safikanov, P. Minakova, S. Novikova, T. Kulova, and A. Yaroslavtsev, Batteries 8[12] (2022) 256.
-
- 44. L. Pang, M. Zhao, X. Zhao, and Y. Chai, J. Power Sources 201 (2012) 253-258.
-
 This Article
This Article
-
2025; 26(3): 377-385
Published on Jun 30, 2025
- 10.36410/jcpr.2025.26.3.377
- Received on Nov 19, 2024
- Revised on Feb 20, 2025
- Accepted on Mar 1, 2025
Services
- Abstract
introduction
experimental
result and discussion
conclusions
- Ethical approval
- Data sharing agreement
- Declaration of Interest
- Funding
- References
- Full Text PDF
Shared
Correspondence to
- Zhuoer Fu
-
School of Engineering and Materials Science, Queen Mary University of London, London, E1 4NS, UK
Tel : +86 15377368635 - E-mail: fuzhuoerdave@gmail.com
Clean-Energy Research Institute(CRI), Hanyang University, 222, Wangsimni-ro, Seongdong-gu, Seoul, 04763, Korea
E-mail: jcpr@hanyang.ac.kr