





- Effects of calcination temperature on properties of sulfuric acid-assisted Y2O3 nano-powder for highly transparent ceramics
Jiao He*, Xin Zhang, Jingbao Lian and Xue Zhang
School of Mechanical Engineering, Liaoning Petrochemical University, Fushun, 113001, P.R. China
This article is an open access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
The transparent Y2O3 ceramic was prepared in this work via vacuum sintering of Y2O3 nanopowder obtained by sulfuric acid-assisted calcination. The calcination temperature notably influenced the dispersion, morphology and sinterability of Y2O3 nanopowders. The calcined powders at various temperatures showed similar shapes, primarily comprised of round particles with an isotropic surface. Nevertheless, the particle size gradually increased as the calcination temperature was raised. The powder calcined at 1100 ℃ showed a lower agglomerate state (n = 2.19) and better sintering properties instead of those obtained at other calcination temperatures, which can be correlated with a larger specific surface area at the low temperature of <1100 ℃ and the rapid growth of large particles at 1200 ℃, respectively. The powder, calcined at 1100 ℃ was vacuum sintered in transparent ceramics for 5 h at a temperature of 1700 ℃. The transparent ceramic exhibited a dense and uniform microstructure, and the 1.5 mm thick sample has an inline transmission of up to 70% at 800 nm wavelength.
Keywords: Transparent ceramics, Y2O3 nano-powders, Calcination temperature, Sulfuric acid.
Transparent polycrystalline Y2O3 ceramics have the remarkable merits of high thermal conductivity (k = 12.8 W/(mK) at 300 K), wide transparency (from 0.2 to 8 µm), a high melting point of 2430 ℃, as well as low phonon energy (592 cm-1), and are thus promising optical materials for transparent media for instance infrared domes and windows, bulb envelopes and laser hosts [1-4]. To easily achieve high transparency and density of polycrystalline Y2O3 ceramics, a key technology is the production of highly sinterable Y2O3 powders that show uniformly dispersed states, ultrafine particles, and narrow particle size distribution (PSD) [5, 6]. Therefore, the research on the production of Y2O3 nanopowders of high quality by a variety of processing techniques has experienced a sharp increase during the past few decades, for instance, precipitation approaches [7-9], hydrothermal treatment [10], sol-gel processing [11], and combustion synthesis [12], etc. Among them, it is generally agreed that the precipitation routes are attractive owing to their feasibility, low cost, and environmental protection features [9].
A large volume of previous works on precipitation methods for synthesizing sinterable Y2O3 emphasized the use of the anionic dispersant containing SO42--ions [13-21]. SO42--ions can adsorb on the particle surface, resulting in the coarsening of particles upon calcination through mass transportation mechanisms. Similar results have been obtained for preparing other oxide ceramic powders added with SO42- using precipitation methods [22-29]. Generally, the properties of Y2O3 powder acquired by the SO42- assisted process rely primarily on two challenging problems. The first problem is the procedure for the formation of the Y2O3 precursors, which is controlled by the optimization of the type of precipitant, concentration of reactants, dosage of the dispersant containing SO42-, reaction time, etc. The second one is the appropriate calcination conditions of SO42- assisted Y2O3 precursors. Besides, its influence mechanism has yet to be confirmed. The main reason may be the desulfurization of sulfate and decomposition of precursor during high-temperature calcination. The continuous occurrence of two thermal decomposition processes makes the determination of the mechanism of calcination conditions on sulfur-doped powder more difficult.
Furthermore, the precipitation method has developed various SO42- assisted Y2O3 precursors (Yttrium carbonate, yttrium hydroxide precipitate, etc.), leading to more difficulty in confirming the appropriate calcination conditions. Qin et al. prepared Y2O3 precursor via the urea precipitation method regulated by (NH4)2SO4. The well-dispersed Y2O3 powders (mean particle size of about 100 nm) were acquired by calcining the precursors at 1100 ℃. The powders were subsequently sintered under a vacuum to produce transparent Y2O3 ceramics [14]. Ikegami et al. prepared the flake-like yttrium hydroxide powder added with 10 mol% of ammonium sulfate. In their work, the mono-dispersed Y2O3 powder was obtained through calcination at 1100 ℃, which was further sintered into transparent ceramic under vacuum at 1700 oC for 5 h [7]. Zhang et al. successfully synthesized hydroxy-carbonate precursors with (NH4)2SO4 by spray coprecipitation. The optimal calcination temperature for high sinterability Y2O3 powders was 1250 ℃ [30].
The calcination temperature substantially impacts the morphology, purity, sinterability, and microstructure of powders [31, 32]. A number of reactions may occur during calcination, including dehydration, the decomposition of intermediate phases, phase crystallization, and, in particular, agglomeration [33]. Nevertheless, very limited efforts have been made to systematically investigate the impacts of calcination temperature on the properties of Y2O3 powder by using SO42- as the regulator, and it still needs to be determined how calcination temperatures affect the morphological and sinterability of the powders. Therefore, finding the desirable calcination conditions for synthesizing Y2O3 nanopowders suitable for fabricating transparent ceramics is of great value. The current work is dedicated to the systematic investigation of the impacts of calcination temperature (900 ℃ to 1200 ℃) on Y2O3 powder particle features by directly pre-treating the Y2O3 powder with sulfuric acid, including morphology property, particle size, degree of agglomeration, and sintering behavior, and based on which to improve the sinterability of the available ceramics powders doped with sulfuric acid. Besides, the sintering behaviors of the powders made from different calcination temperature are also evaluated by scrutinizing the microstructure and optical properties of the resultant Y2O3 ceramics produced by vacuum sintering at 1700 oC for 5 h.
Preparation process
A commercial Y2O3 powder (with a purity of 99.99%; Huizhou Ruier Rare Chemical Hi-Tech Co., Ltd, Huizhou, China) was utilized as a raw material. The mean specific surface area of the powder is approximately 31.5 m2·g-1
(as identified with BET measurements). The powder was dispersed in ethanol, and 7 mol% sulfuric acid was added (the dosage of sulfuric acid were given as a molar proportion of the Y2O3 powder basic (mol%)). Then the suspension was ball-milled with ZrO2 balls for 8 h to break up the agglomerate. The suspension obtained was dried in an oven at 110 ℃ and then sieved by a nylon sieve of 200 mesh. Subsequently, the powder was calcined in the air from 900 ℃ to 1200 ℃ for 4 h, with a heating rate of 10 ℃·min-1 applied. Lastly, the calcined powders were dry-pressed in a die of 15 mm at 100 MPa pressure with a subsequent cold isostatic pressing (CIP) at 200 MPa. In a vacuum furnace (VSF-7, Shenyang, China), the resulting green compacts were sintered with a molybdenum heating element at 1700 ℃ for 5 h under vacuum. The pressure was less than 10-3 Pa.
Characterization
The morphologies of the powders were examined by High-resolution transmission electron microscopy (TEM, Model JEM-2100F, JEOL, Tokyo, Japan). The average particle size (DTEM) was examined from more than 200 randomly chosen particles from the images of TEM. The specific surface area of the powders was measured with BET (Model TriStar II 3020, Micrometritics Instrument Corp., Norcross, GA), and nitrogen was applied as an absorbate. The specific surface area of the powders was converted into particle size (DBET) in accordance with the below equation [26]:

Here, ρ represents the theoretical density of the Y2O3 crystal (which is 5.01 g·cm-3); S represents the specific surface area that was determined by BET measurement.
The phase identification was carried out through X-ray diffractometry (XRD, Model PW3040/60, Philips, Eindhoven, The Netherlands) utilizing the graphite monochromatic CuK radiation (with λ of 0.15406 nm) at 40 kV/40 mA. For the calcined Y2O3 powders, the mean grain size (DXRD) was estimated by applying a peaking broadening approach for the (222) diffraction of Y2O3, which was derived from the Scherrer equation:

Here, λ represents the incident X-ray wavelength (0.15406 nm); θ represents the diffraction angle and β denotes the width of the diffraction line at half intensity, in radians.
By the dilatometry method on the thermodynamic analyzer (SETSYS Evolution 2400, Setaram, Caluire, France), the densification behavior of powder compacts was examined in 20 ml·min-1 flowing oxygen, while maintaining constant heating and cooling rates of 10 and 40 ℃·min-1, respectively.
The densities of the green compacts were measured by the gravimetric method, and the density of the vacuum-sintered samples was determined utilizing the Archimedes method in water. The relative density was subsequently calculated by comparing the actual density of Y2O3 to its theoretical density.
The optical transmittance of samples (with a thickness of 1.5 mm) that were polished on two sides was determined in the 200-2000 nm wavelength with a ultraviolet/visible/near infrared spectrophotometer (Model Lambda 750S, Perkin Elmer, CT, USA). A scanning electron microscope (SEM, Model JSM-7001F, JEOL, Tokyo, Japan) was employed for viewing the microstructure of sintered ceramics.
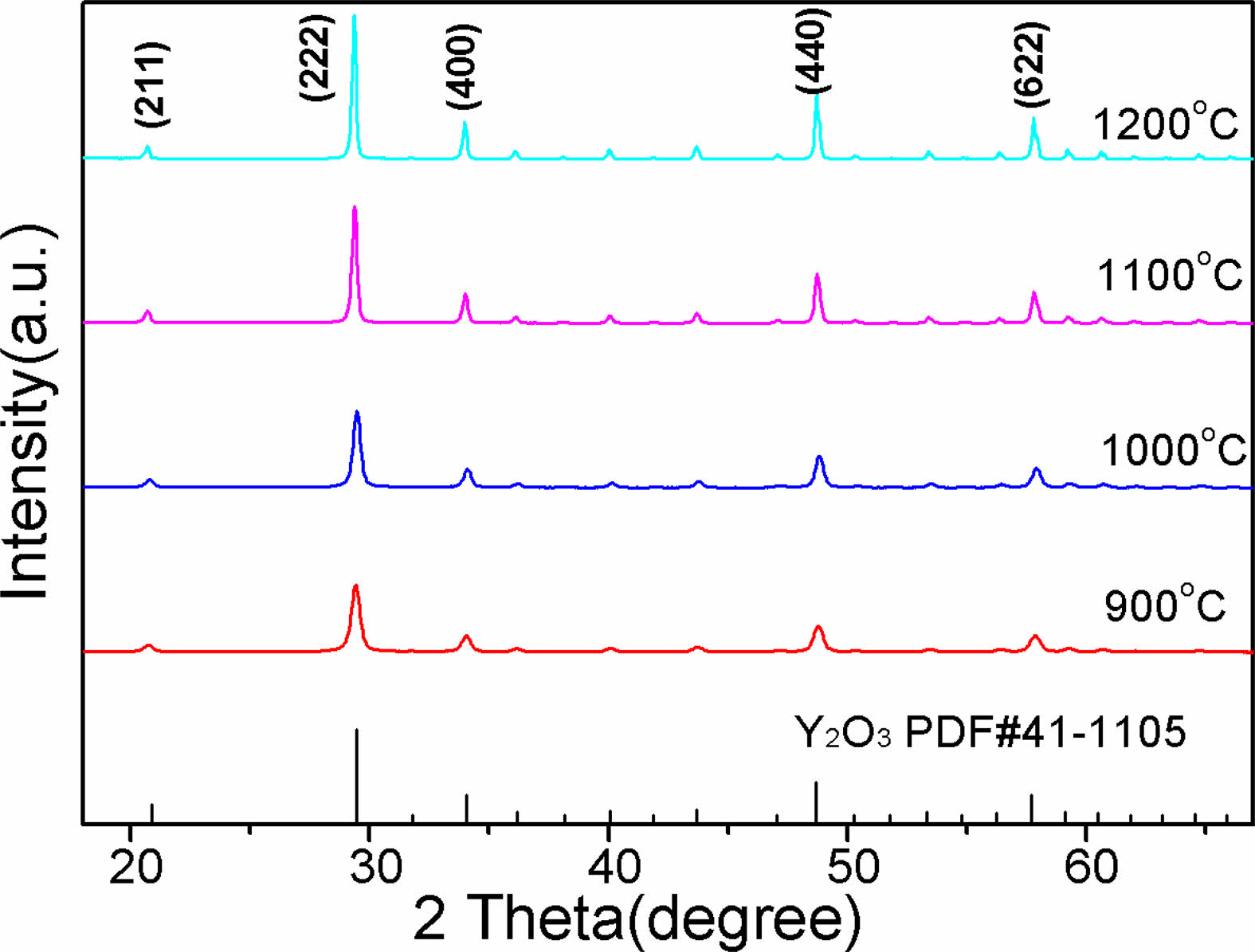
Fig. 1 displays the XRD patterns of the Y2O3 powders that were calcined at various temperatures for 4 h, and as can be observed, all the characteristic diffraction peaks of the calcined powders are well indexable with the standard phase of the cubic Y2O3 (JCPDS #41-1105). A steady rise in the intensity of the characteristic diffraction peaks was observed at higher calcination temperatures. In contrast, the full-width at half-maximum (FWHM) of each peak got narrower, suggesting that a larger grain size and better crystallinity were reached.
Fig. 2 presents the TEM images of the Y2O3 powders obtained by calcining the sulfuric acid pre-treated Y2O3 at various temperatures for 4 h. It is noticed that the calcination temperature had a marked impact on the morphology, agglomeration state, and particle size of the products. All the Y2O3 powders were composed of round particles with isotropic surfaces and quite similar shapes. The average particle size of Y2O3 powder estimated from observation of TEM rised in the range of 35-120 nm as the calcination temperature increased in a range of 900 ℃-1200 ℃. At calcination temperatures below 1100 ℃ as shown in Fig. 2(a-b), severe agglomeration occurred between particles owing to their relatively smaller particle sizes and higher surface energy [30]. At a calcination temperature of 1200 ℃, the sintering necks between adjacent particles were generated due to the faster mass transfer and diffusion. as a consequence, more serious agglomeration happened (Fig. 2(d)). It is noteworthy that the hard aggregates produced at 1200 ℃ were extremely difficult to be broken up during the powder processing stage, while the powders calcined at 1100 ℃ showed no apparent sintering necks between particles and excellent homogeneity on a large scale, indicating a rather loosely agglomerated state. Through dry pressing and cold isostatic pressing, the weak aggregation can be eliminated easily during the powder consolidation phase, where deformation and plastic flow would take place to fill the spaces among the particles and correspondingly acquire a green compact with homogeneous microstructure[30].
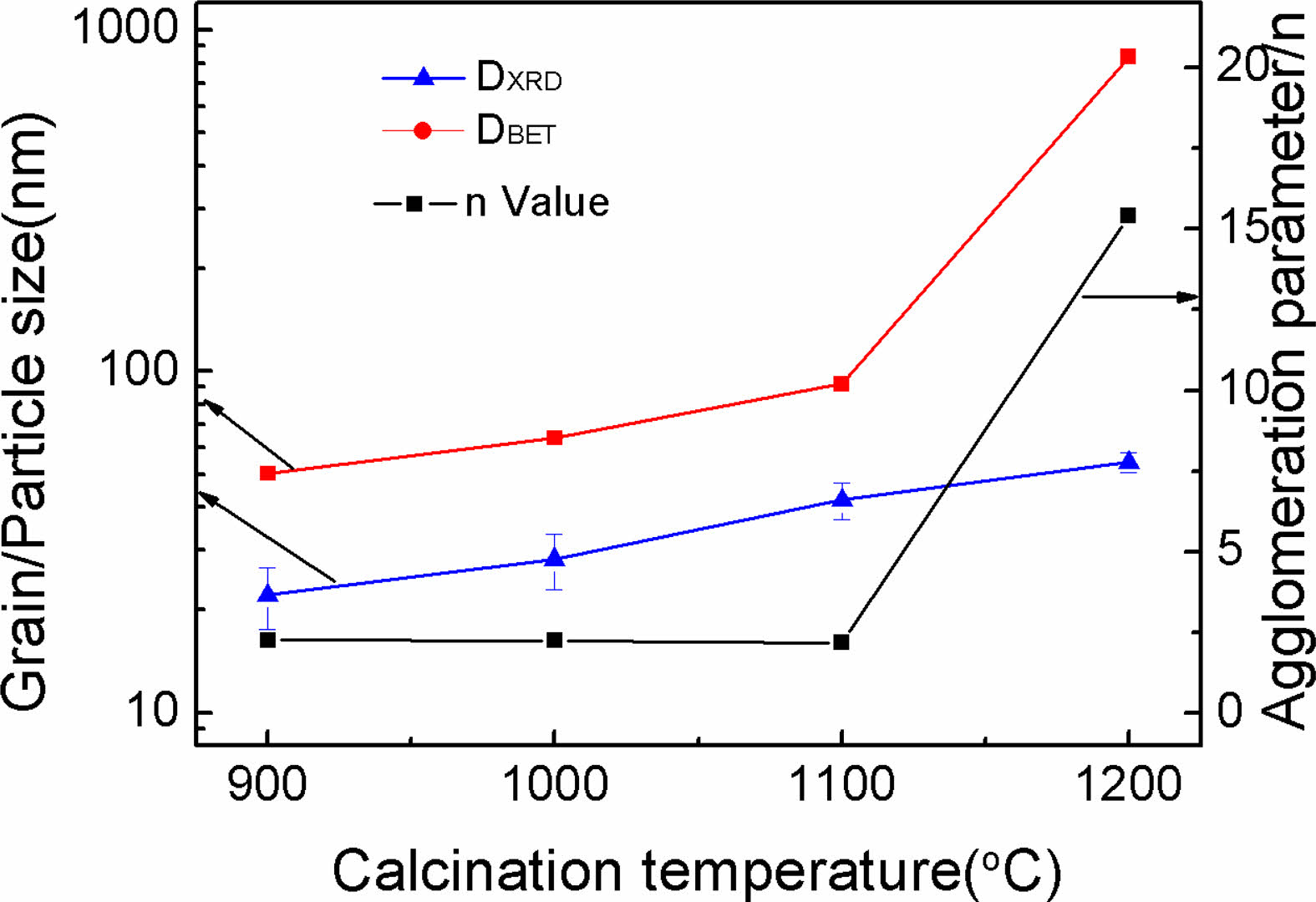
The particle sizes of Y2O3 powders determined via BET measurement (DBET) and the mean grain sizes obtained by peaking broadening analysis (DXRD) are listed in Fig. 3. The increased tendencies of both DBET and DXRD with increasing calcination temperature are depicted in Fig. 3. It is well established that the degree of difference between DBET and DXRD offered evidence revealing the particle agglomeration state. Accordingly, the calculated ratios of DBET/DXRD (n) for the powders are also shown in Fig. 3. The n value slightly reduced when the calcination temperature was below 1100 ℃, while a significant increase happened at the higher temperature of 1200 ℃, indicating that well dispersity and favorable crystallinity of the Y2O3 powder can be simultaneously achieved at 1100 ℃ (n = 2.19). The n value reached the high level of around 15 at 1200 ℃, which was mainly due to occurrence of hard aggregation. The above phenomenon agrees well with the morphology observation results (Fig. 2).
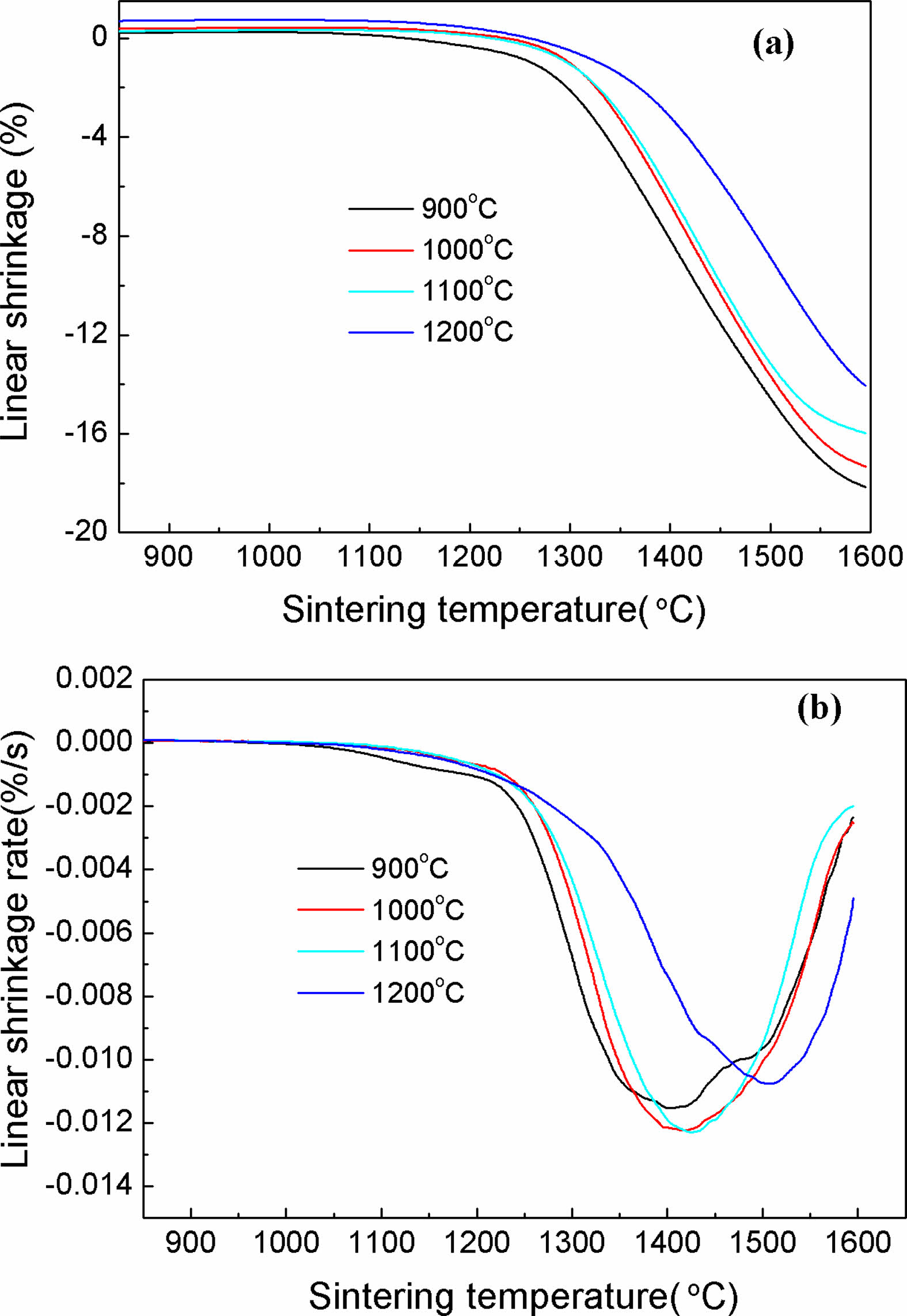
The impacts of calcination temperatures on the sintering behavior of calcined Y2O3 powders at 900, 1000, 1100 and 1200 ℃ were examined with a thermal mechanical analyzer (TMA). The results are presented in Fig. 4. The green compacts made from these powders were calculated to have relative densities of about 48.93, 50.29, 53.09 and 54.28%, respectively. Fig. 4(a) illustrates the shrinkage behaviors of these Y2O3 powders during heating at 10 ℃·min-1, where no obvious shrinkage was observed for all the powders when the calcination temperature was below 1150 ℃. Afterward, apparent shrinkage occurred from ~1200 to 1600 ℃, leading to the total shrinkage of the powders calcined at 900, 1000, 1100, and 1200 ℃ at 18.15, 17.34, 15.99, and 14.07% respectively. The powder obtained at a lower calcination temperature showed a more obvious shrinkage, which was ascribed to the faster speeding-up material transport for the powders with smaller particle sizes[7]. The shrinkage curve in Fig. 4(a) was utilized to identify the densification rate of the powders as a function of temperature, as exhibited in Fig. 4(b). All samples displayed a broad peak with the valley point centered at sintering temperature range of 1400 to 1550 ℃, based on which a marked densification temperature can be estimated. It exhibits that the samples calcined at 1200 ℃, different from the others, exhibited the highest sintering temperature of pronounced densification above 1500 ℃, indicating a decreased sinterability. The 1100 ℃ calcined powder showed the temperature of pronounced densification of about 1425 ℃, with a shrinkage rate of 0.0124 %·s-1.
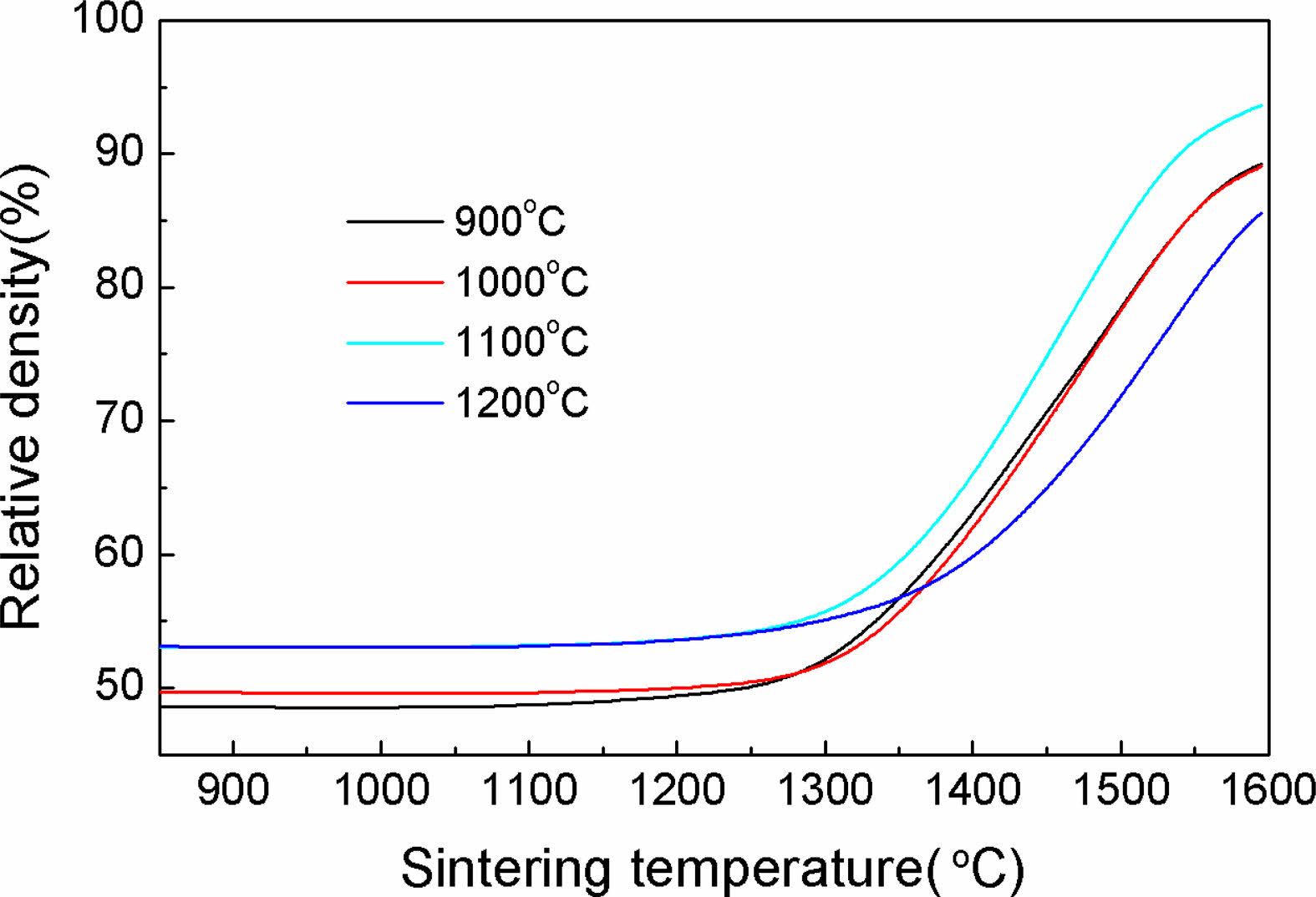
Fig. 5 displays the temperature dependence of the density for all powders, the density of the powder compact ρ,at any temperature, is determined from the green density ρ0 and the calculated linear shrinkage ∆L/L0, using the following equation[34]:

In which L0 and L denotes the initial and instantaneous lengths of the sample, respectively. ∆L = L-L0. It can be noted that within the selected range of calcination temperatures, the densification of Y2O3 powder was significantly enhanced. Raising the calcination temperature can effectively contribute to the densification of Y2O3 powder, and the final density for the sample calcinated at 1100 ℃ reached 93.63% at the sintering temperature of up to approximately 1600 ℃. Further raising the calcination temperature to 1200 ℃ does not yield any additional benefits, but instead slowed densification, leading to a reduction in final density to 85.56%. This is supported by the results displayed in Fig. 4.
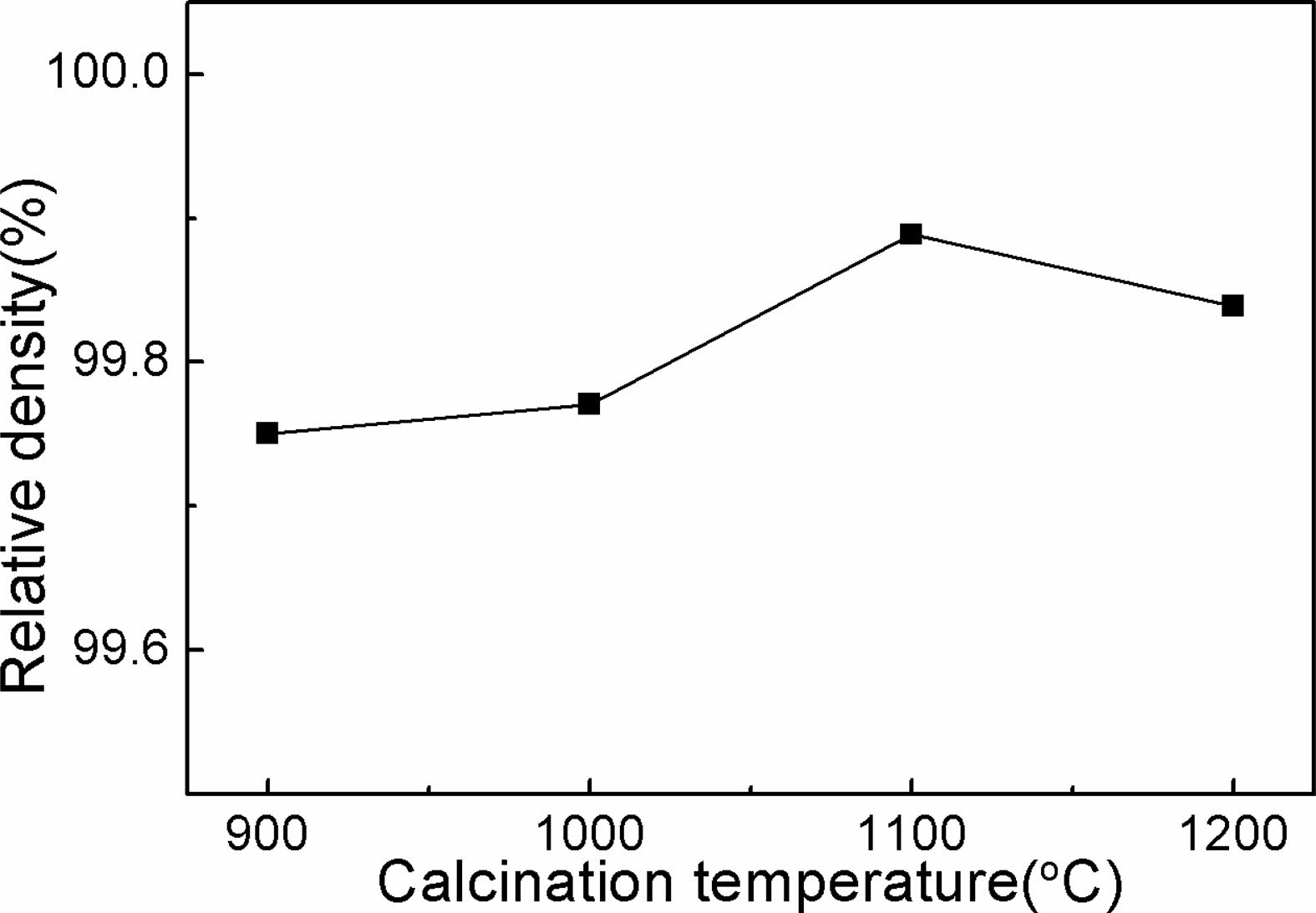
For further assessment of the sintering behavior of the powders, all the samples were sintered at a temperature of 1700 ℃ for 5 h under vacuum. Fig. 6 illustrates the relative densities of Y2O3 ceramics as a function of the powder calcination temperature. It is evident that all samples realized a marked densification after vacuum-sintering. The Y2O3 powders calcined at 1100 ℃ were almost sintered into transaprent ceramic with high density (above 99.89%), and Y2O3 ceramics derived from the other powders also reached densities between 99.75% and 99.84%. The average particle sizes of calcined samples at 900 ℃ and 1000 ℃ were relatively smaller than those obtained at higher temperatures, which endowed the powders with larger surface area, as shown in Fig. 3. The high friction between the die and the small particles, as well as between the small particles, makes it difficult to acquire green compacts with good uniformity and high density during dry compaction, and therewith, the as-obtained green compacts showed poorer sinterability than that made of the 1100 oC calcined powder. As for calcined powders at a higher calcination temperature (1200 ℃), sintering necks and significant increase in particle size happened, leading to the reduced surface free energy of particles, which further caused a weak densification driving force and poor sintering activity[35]. Therefore, a calcination temperature of 1100 ℃ is crucial to achieve a Y2O3 powder with excellent dispersion and high sinterability.
Besides, notably, ceramic samples produced from the calcined powder at 1200 ℃ showed higher relative density than those made from the calcined powders at lower temperatures (900 ℃ and 1000 ℃). This phenomenon seems to be inconsistent with the sintering behaviors of the powders investigated in Fig. 5, since the higher sintering temperature of 1700 ℃ may have enhanced the sintering driving force, and thus, leading to further densification for the Y2O3 powder with larger particle size.
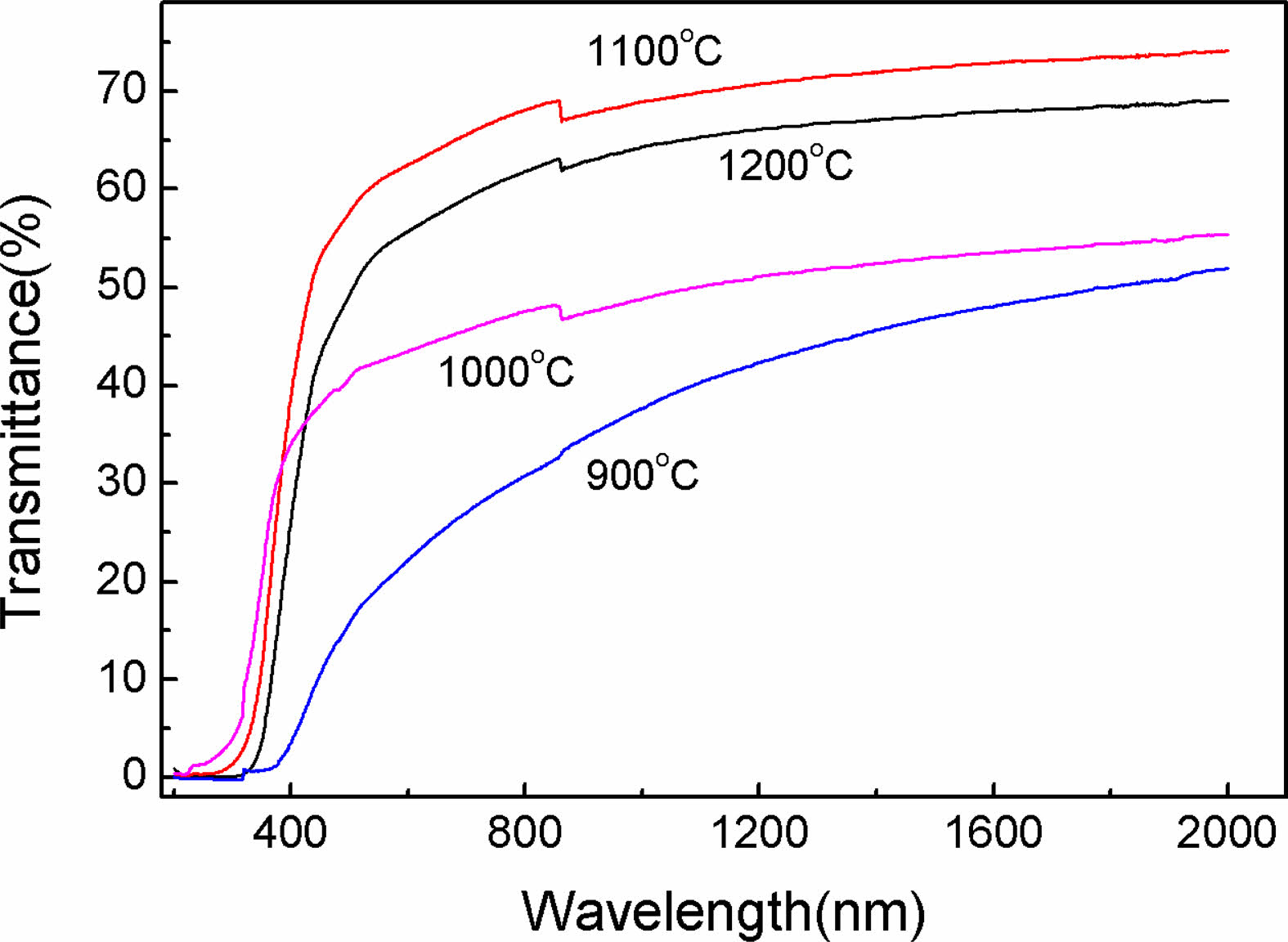
After vacuum sintering and fine polish, the Y2O3 transparent ceramics (1.5 mm-thick) from the powders calcined at diverse temperatures were obtained, and the corresponding photographs are displayed in Fig. 7. It is evident that all the ceramics were extremely transparent. The marks underneath the samples were clearly visible. A comparison of the in-line transmittance of ceramics is given in Fig. 8. Notably, the transmittance rised noticeably with rising calcination temperature, reaching a maximum of around 70% at 800 nm for ceramics derived from powders calcined at 1100 ℃, indicative of its good sintering activity. Further, an increase in powder calcination temperature to 1200 ℃ did not contribute to a further increase in the optical transmission of the ceramics.
Fig. 9 compares the microstructure of the fracture surfaces of gold-coated ceramics from powders calcined at different temperatures. It is evident that the Y2O3 ceramics showed a rise in grain size with an increase in calcination temperature in a range of 900 ℃-1200 ℃, which was caused by the larger particle obtained from higher calcination temperature. These surfaces also indicated that the Y2O3 ceramics made from the calcined powders at 1100 ℃ and 1200 ℃ possessed an almost pore-free, dense structure (Fig. 9c, d), whereas the ceramics made from the powders calcined at lower temperatures showed a small quantity of tiny pores in the grain and at the boundary, as denoted by the red circles (Fig. 9(a, b)). Our previous research has established that doping of sulfate assists the mechanism that results in coarsening of the particles at temperatures <1250 ℃, generating sinterable powder and loose agglomerate. As a result, compared with the sulfate-absent ceramics [36], all the ceramics made from the powders doped with sulfate and calcined at different temperatures in the present work had higher densities and more homogeneous microstructures. Besides, a calcination temperature of less than 1100 ℃ was not obviously high enough to induce massive decomposition of Y2(SO4)3 in the powders [36]. The product after calcination comprised particles from a mass of Y2O3 and a small number of Y2O2SO4 particles. Therefore, the complex composition and severe aggregation of powders calcined at 900 ℃ and 1000 ℃ (Fig. 2) resulted in a relatively poor sinterability, causing residual porosity in the resultant ceramics. For powders calcined at 1200 ℃, the large particle size excessively reduced densification driving force. Thus, it is unambiguously said that the most appropriate calcination temperature for the sulfate-doped Y2O3 powder used for fabrication of transparent Y2O3 ceramic was 1100 ℃.
|
Fig. 1 XRD patterns of Y2O3 powder that was calcined at different temperatures. |

|
Fig. 2 TEM micrographs of calcined Y2O3 powders at diverse temperatures, namely, 900 ℃ (a), 1000 ℃ (b), 1100 ℃ (c), and 1200 ℃ (d). |
|
Fig. 3 The n-values and particle sizes for the calcined powders at a variety of temperatures for 4 h. |
|
Fig. 4 (a) Shrinkage and (b) shrinkage rate curves for the Y2O3 powders calcined at different temperatures in the course of heating. |
|
Fig. 5 Density evolution for the Y2O3 powders calcined at various temperatures as a function of sintering temperature. |
|
Fig. 6 Densification behaviors of the Y2O3 ceramics made from the calcined powders at various temperatures. |

|
Fig. 7 Photos of the Y2O3 polished ceramics from powders that were calcined at a variety of temperatures. |
|
Fig. 8 Transmittance spectra of Y2O3 ceramics produced from powders that were calcined at various temperatures. |

|
Fig. 9 SEM micrographs of the Y2O3 ceramic fracture surfaces produced from powders calcined. The red circles in (a) and (b) indicate tiny poles. 900 ℃ (a), 1000 ℃ (b), 1100 ℃ (c), and 1200 ℃ (d). |
Transparent Y2O3 ceramic was prepared via vacuum sintering using nano-sized and sulfuric acid-containing Y2O3 powder calcined at an appropriate temperature. The effect of calcination temperature on morphology, dispersion, and sinterability of Y2O3 nanopowders was systematically examined. After calcination, all powders were composed of isotropic, round particles. Additionally, the particle size of Y2O3 powder progressively increased with an increase in calcination temperature. Sintering behaviors for Y2O3 powders showed that the lower calcination temperatures (900 ℃ and 1000 ℃) gave rise to larger surface area and size of agglomerate, causing a reduction in the sinterability of the powders. On the other hand, the higher calcination temperature of 1200 ℃ triggered the growth of the large particles and the formation of sintering neck, which were unfavorable for densification. The powder had the best dispersive properties with the smallest ratio of DBET/DXRD (n) and excellent sinterability when calcined at 1100 ℃ for 4 h, and the corresponding sintered ceramic with a relative density of ~99.89% was fabricated by vacuum sintering at 1700 ℃ for 5 h. It showed an in-line transmittance of 70% at 800 nm.
This work was supported by the National Natural Science Foundation of China (Grant No. 51802136), Foundation of Liaoning Educational Committee (Grant No. L2020043).
- 1. H. Furuse, S. Nakasawa, H. Yoshida, K. Morita, T.S. Suzuki, B.N. Kim, Y. Sakka, and K. Hiraga, J. Am. Ceram. Soc. 101[2] (2018) 694-702.
-
- 2. C.R. Liu, J. Jiang, G.H. Zhou, T.J. Zhang, S. Hu, L. Gan, and X.P. Qin, J. Am. Ceram. Soc. 105[5] (2022) 3382-3390.
-
- 3. Y.G. Yanga, J.Y. Kwakb, H. Kongc, and S.J. Lee, J. Ceram. Process. Res. 21[4] (2020) 450-455.
-
- 4. C.W. Park, J.H. Lee, S. H. Kang, J.H. Park, H.M. Kim, H.S. Kang, H.A. Lee, J.H. Lee, J.H. In, and K.B. Shim, J. Ceram. Process. Res. 19[2] (2018) 179-182.
-
- 5. J.G. Li, T. Ikegami, T. Mori, and Y. Yajima, J. Am. Ceram. Soc. 87[6] (2004) 1008-1013.
-
- 6. X. Zhang, W. Lu, G. Fan, and X. Wang, Adv. Powder Technol. 27[1] (2016) 295-298.
-
- 7. T. Ikegami, J.G. Li, T. Mori, and Y. Moriyoshi, J. Am. Ceram. Soc. 85[7] (2002) 1725-1729.
-
- 8. M. Kabir, M. Ghahari, and M. Afarani, Ceram. Int. 40[7] (2014) 10877-10885.
-
- 9. S.S. Li, B.L. Liu, J. Li, X.W. Zhu, W.B. Liu, Y.B. Pan, and J.K. Guo, J. Alloys. Compd. 678 (2016) 258-266.
-
- 10. M. Ghaderi, R.S. Razavi, M.R. Loghman-Estarki, and S. Ghorbani, Ceram. Int. 42[13] (2016) 14403-14410.
-
- 11. S.Q. Xu, J. Li, C.Y. Li, Y.B. Pan, and J.K. Guo, J. Am. Ceram. Soc. 98[9] (2015) 2796-2802.
-
- 12. R.V. Mangalaraja, J. Mouzon, P. Hedström, Carlos P. Camurri, S. Ananthakumar, and M. Odén, Powder Technol. 191[3] (2009) 309-314.
-
- 13. H.M. Qin, H. Liu, Y.H. Sang, Y.H. Lv, X.L. Zhang, Y.Y. Zhang, T. Ohachic, and J.Y. Wang, Cryst Eng Comm. 14[5] (2012) 1783-1789.
-
- 14. H.M. Qin, X.H. Zhang, H. Liu, Y.H. Sang, and J.Y. Wang, Cryst Eng Comm. 15[25] (2013) 5076-5081.
-
- 15. Y.F. Liu, X.Y. Qin, H.X. Xin, and C.J. Song, J. Eur. Ceram. Soc. 33[13-14] (2013) 2625-2631.
-
- 16. N. Saito, S. Matsuda, and T. Ikegami, J. Am. Ceram Soc. 81[8] (1998) 2023-2028.
-
- 17. T. Onodera, T. Ikegami, Y. Yajima, M. Kawamura, M. Sakai, and Y. Moriyoshi, J. Ceram. Soc. Jpn. 111[9] (2003) 664-668.
-
- 18. L. Wen, X.D. Sun, Z.M. Xiu, S.W. Chen, and C.T. Tsai, J. Eur. Ceram. Soc. 24[9] (2004) 2681-2688.
-
- 19. L. Wen, X.D. Sun, Q. Lu, G.X. Xu, and X.Z. Hu, Opt. Mater. 29[2-3] (2006) 239-245.
-
- 20. J. Li, W.B. Liu, B.X. Jiang, J. Zhou, W.X. Zhang, L. Wang, Y. Shen, Y. Pan, and J. Guo, J. Alloys. Compd. 515 (2012) 49-56.
-
- 21. Z.G. Huang, X.D. Sun, Z.M. Xiu, S.W. Chen, and C.-T. Tsai, Mater. Lett. 58[15] (2004) 2137-2142.
-
- 22. F. Kara and G. Sahin, J. Eur. Ceram. Soc. 20[6] (2000) 689-694.
-
- 23. J.G. Li, T. Ikegami, T. Mori, and Y. Yajima, J. Mater. Res. 18[5] (2003) 1149-1156.
-
- 24. Y.H. Lv, W. Zhang, H. Liu, Y.H. Sang, H.M. Qin, J. Tan, and L.N. Tong, Powder Technol. 217 (2012) 140-147.
-
- 25. X.J. Xu, X.D. Sun, H. Liu, J.G. Li, X.D. Li, D. Huo, and S.H. Liu, J Am Ceram Soc. 95[12] (2012) 3821-3826.
-
- 26. X.X. Li and W.J. Wang, Powder Technol. 196[1] (2009) 26-29.
-
- 27. J. Li, J.P. Li, Q. Chen, W.J. Wu, D.Q. Xiao, and J.G. Zhu, Powder Technol. 218 (2012) 46-50.
-
- 28. J.Q. Wang, S.H. Zheng, R. Zeng, S.X. Dou, and X.D. Sun, J. Am. Ceram. Soc. 92[6] (2009) 1217-1223.
-
- 29. H.L. Li, X.J. Liu, and L.P. Huang, Ceram. Int. 32[3] (2006) 309-312.
-
- 30. L. Zhang, Z. Li, F.Z. Zhen, L.X. Wang, Q.T. Zhang, R. Sun, F.A. Selim, C.P. Wong, and H. Chen, J. Mater. Sci. 52 (2017) 8556-8567.
-
- 31. Z.H. Ding, W. Zheng, F. Chen, I.J. Kim, W.L. Liu, J.F. Shackelford, H.W. Song, T. Nakayama, S. Kim, Y.-H. Han, and J. Kim, J. Ceram. Process. Res. 24[4] (2023) 675-682.
-
- 32. C.H. Wang, W. Yang, Z.Q. Wang, B. Liu , S.H. Li, L. Tao, X.R. Li , W.P. Miao, and W. Luo, J. Ceram. Process. Res. 24[2] (2023) 374-378.
-
- 33. X.Y. Huang, X. Zhang, Z.W. Hu, Y.G. Feng, J.B. Wei, X. Liu, X.Y. Li, H.H. Chen, L.X. Wu, H.M. Pan, and J. Li, Opt. Mater. 92 (2019) 359-365.
-
- 34. X.C. Song, J. Lu, T.S. Zhang, and J. Ma, J. Am. Ceram. Soc. 94[4] (2011) 1053-1059.
-
- 35. J.S. Li, X.D. Sun, S.H. Liu, X.D. Li, J.G. Li, and D. Huo, Ceram Int. 41[2] (2015) 3283-3287.
-
- 36. J. He, X.D. Li, S.H. Liu, Q. Zhu, J.G. Li, and X.D. Sun, J. Eur. Ceram. Soc. 35[8] (2015) 2369-2377.
-
 This Article
This Article
-
2024; 25(1): 144-150
Published on Feb 29, 2024
- 10.36410/jcpr.2024.25.1.144
- Received on Jan 15, 2024
- Revised on Feb 4, 2024
- Accepted on Feb 15, 2024
Services
- Abstract
introduction
experimental procedure
results and discussion
conclusions
- Acknowledgements
- References
- Full Text PDF
Shared
Correspondence to
- Jiao He
-
School of Mechanical Engineering, Liaoning Petrochemical University, Fushun, 113001, P.R. China
Tel : +86-24-56865042 Fax: +86-24-56865042 - E-mail: hejiao@lnpu.edu.cn
Clean-Energy Research Institute(CRI), Hanyang University, 222, Wangsimni-ro, Seongdong-gu, Seoul, 04763, Korea
E-mail: jcpr@hanyang.ac.kr