





- Geopolymerisation of un-calcined kaolin by the novel heat curing method
Selin Demir and Süleyman Yaşın*
Karabük University, Department of Metallurgy and Materials Engineering, 78050, Karabük, Turkey
This article is an open access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
In this study, heat curing technique (up to 320 oC) that allows the usage of sodium silicate solutions with un-calcined kaolins to offer a durable geopolymeric binder in a short time was investigated. Samples containing up to 40 wt.% binder have been produced to observe the binder behaviour over a wide range of additions by heat curing for one hour. The cured samples consist of kaolinite and amorphous phase and crystallinity was decreased with increasing curing temperatures. 240 oC was the threshold temperature for the strength development of the samples. Condensation reactions between Al-OH- units in un-calcined kaolin and silanol groups in sodium silicate solution that yield sialate (Si-O-Al-O) bridges detected by infrared analyses are the considered mechanisms of promoter effects of kaolinite. Optimum compressive strength (23.4 MPa.) was obtained with the samples produced with 20 wt.% binder addition and 300 oC curing temperature
Keywords: Kaolinite, Geopolymer, Sodium silicate, Heat curing
Geopolymers are inorganic materials commonly pro- duced by the alkalination of alumina silicates at low temperatures [1, 2]. They have applications such as greener cements, ceramics, bricks, refractories, insulation foams and composites [1, 3-7]. In their production, metakaolin produced by the calcination of natural kaolin is normally used as a solid alumina silicate precursor due to its high reactivity [8-10]. The calcination tem- perature of kaolin is a parameter that has important effects on the geopolymer properties [11]. Elimbi et al. reported that the best calcination temperature of kaolin for geopolymer cements is around 700 oC [11]. Heating rate applied to calcination temperature affects mechanical properties and setting times of geopolymers [12]. Other parameters also have effects, such as changing the water demand of the mixtures due to the different morphologies of metakaolin as a result of the calcination with different kiln technologies [13]. Furthermore, unlike metakaolin calcined in a rotary kiln, metakaolin calcined in multiple hearths calciner and flash calciner providing the desired chemical reactivity (Al in 5-fold coordination) [14]. Although kaolin is a natural and common raw material, the specificity of the manufac- turing process of metakaolin for geopolymers decreases the availability of the suitable precursors on the market. In addition, the calcination leads to energy consumption, time loss and CO2 emission.
Some promising studies have been published related to the use of un-calcined kaolin in the production of geopolymers to eliminate calcination [15-17]. Heah et al. cured kaolin-based geopolymers activated with sodium silicate and sodium hydroxide solutions at 80 oC [15]. The optimum compressive strength was reported as 5.75 MPa for the samples cured for three days [15]. Slaty et al. achieved compressive strengths up to 48 MPa (48-hour curing at 80 oC) with kaolin-based geopolymers moulded with a pressure of 16 MPa [16]. Hounsi et al. reported that the compressive strength increased in final samples with mechanical activation of kaolin in a planetary mill [17]. In the studies on synthesis of geopolymer with un-calcined kaolin, reaction kinetics were observed lower than those of metakaolin-based geopolymers [15, 16, 18].
Transparent and amorphous silica based solid materials were obtained just by drying pure sodium silicate solutions [19]. The negative side of dried (heat cured) sodium silicate binder is that they lose their strength by atmospheric moisture [20]. Sodium silicate solutions are commonly used in geopolymer formula- tions to obtain high performance products [21, 22]. In contrast to heat curing of sodium silicate binder alone, geopolymeric reactions offer chemically stable cross-linked materials [9, 23, 24]. Geopolymeric modification of sodium silicate binder by incorporating sodium aluminate solution have successful applications in metal castings [25, 26]. Another developed method involves mixing sodium silicate solution, sand and un-calcined kaolin then heating the mix between 200 - 300 oC [27]. Available bonded water in un-calcined kaolin supported the polycondensation between kaolin and sodium silicate solution [27]. The condensation reactions gave a geopolymeric character to the ordinary sodium silicate binder and led to higher strength and enhanced humidity resistant by incorporating alumina silicates into the silica-rich framework [27]. The binder has application potential as a more durable alternative to pure sodium silicate solution where the heat curing applied. The heat curing method also offers an alterna- tive way to produce un-calcined kaolin based geopoly- mers in a shorter time, but process parameters and cured properties are not clear enough. In this study, it is aimed to characterise the kaolin based geopolymer produced with the heat curing process by systematic investigations.
Materials
Kaolin powders with a particle size of D(0.5)=0.87 µm were used. The chemical analyses of the kaolin are given in Table 1. For the preparation of the liquid part, sodium silicate solution (Merck) in a composition of 27 wt.% SiO2, 8 wt.% Na2O and 65 wt.% H2O, sodium hydroxide pellets (Merck) in 99 wt.% purity and distilled water were used. EN 196-1 CEN standard sand was used as a filler.
Sample preparation
The most preferably molar ratio of Na2O/Al2O3 was reported 1.1 to 0.9 and SiO2/Al2O3 was 4 to 5.5 for this type of heat curable geopolymeric binder formulation [27]. The molar composition of the binder produced in the study was kept constant as Na2O/Al2O3=1, SiO2/Al2O3=5 and H2O/Al2O3=30 since high strength with good workability was obtained in the preliminary tests with this composition [28]. The liquid part was pre- pared by adding water and NaOH pellets to the sodium silicate solution to provide molar ratios. The resulting solution was mixed with the appropriate proportions of sand using an electric mixer for 120 sec. Kaolin powders were added to the prepared mixture and final mixing was applied for 120 sec. Mixtures were obtained by adding different amounts of binder (the total mass of the binder is equal to the sum of the liquid part and the kaolin) ranging from 2.5 to 40 wt.% to the sand. The wet mixtures obtained were placed in 50 mm x 50 mm x 50 mm steel cube moulds. After the surfaces were levelled with a trowel, the shaped samples were removed from the mould. Two-step heat treatment was applied to avoid observed deformation (foaming) in one-step heat treatment trials for temperatures 260 oC or more. In the first step demoulded samples were placed in an oven preheated to 240 oC for 30 min. In the second step, samples heated from 240 to targeted temperatures with a heating rate of 20 oC/min. and samples heat cured at those temperatures for another 30 min (for the samples prepared for 200 and 240 oC, heat curing was applied in one step for 60 min. due to lower temperatures). For the analyses of FT-IR (Fourier-trans- form infrared spectroscopy), XRD (X-ray diffraction), and DTA-TG (differential thermal analysis and ther- mogravimetry) samples from pure binder were synthesised with the same method without sand addition to obtain clear results.
Characterisation
The compressive strength tests applied to the samples were carried out in a Zwick/Roell 600 kN universal mechanical test device. The testing rate was 1mm/min. Maximum strength before breaking was recorded for each sample. The average of three samples was recorded as the compressive strength value. To examine the phase structure of the samples, XRD analysis was performed at a rate of 5o/min. in the range of 5-70o. Thermal analysis of the samples was conducted in a Hitachi STA 7300 DTA-TG device. DTA-TG analysis was per- formed at temperature ranges between room tempera- ture and 700 oC with a heating rate of 5 oC/min in a nitrogen atmosphere. The bond structure of the samples was examined in the transmittance mode with a Bruker FT-IR device in the range of 400-4000 cm-1. Micro- structural investigation of the samples was carried out with a Carl Zeiss Gemini FE-SEM (Field emission scanning electron microscopy) device.
Appearance of the samples
While the samples did not show deformation up to 20 wt.% binder addition (Fig. 1a-d), deformation that can be defined as expansion occurred in the samples produced with the addition of 30 and 40 wt.% binders (Fig. 1e, f). The synthesized geopolymers by alkalina- tion of kaolin [29, 30] or silica fume [31] without soluble silicates (only with alkali hydroxides) was not shown this type of dramatic expansions in the previous studies. On the other hand, significant expansion was reported for the geopolymers produced with alkali silicate solution and fumed silica [31]. Polycondensation of silanol groups into siloxo generate H2O above 200 oC [32]. The polycondensation water may lead to expansion of the samples include 30 and 40 wt.% binder due to insufficient permeability to drain the water vapour.
Compressive strengths of the samples
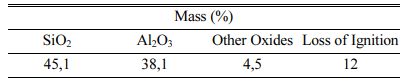
In Fig. 2, compressive strengths of the samples with 20 wt.% binder addition at different curing temperatures are given. The sample cured at 200 oC showed the lowest compressive strength as 1.29 MPa. The low compressive strength may be related to insufficient dehydration of the samples at the applied curing temperature and time. The hygroscopic nature of the binder (due to kaolin and sodium-rich alkaline activator) with other parameters such as binder dosage, and the geometry of the samples may lead to delayed drying and condensation reactions. The semi-wet character of these cured samples was also observed by the naked eye with their darker colours. The compressive strength reached 19.5 MPa after curing at 240 oC. This significant increment indicates that 240 oC is a threshold temperature for the sufficient dehydration and possible condensation reactions occurring in the binder. Lucas et.al. investigated the interaction between silica sand and sodium silicate solution during consolidation by drying at different temperatures [33]. They reported that at 70 oC drying temperature, sodium silicate acts as a binder for sand grains [33]. Dissolution-precipitation reaction occurred in the mixture consoli- dating more strongly at 150 oC drying temperature [33]. Free sodium ions increase the reactions between sodium silicate solutions and quartz sand surface [25]. On the other hand, it is reported that the geopolymeric modifications (reactions) binds sodium ions in sodium silicate solutions [25]. This situation indicates that the strength enhancement with increasing temperature pro- bably originated from the interactions between sodium silicate and kaolin together with interactions between sodium silicate and sand in the produced samples. The highest compressive strength was measured as 23.4 MPa for the sample cured at 300 oC. There is a gradual dehydration in geopolymer material with increasing temperature [34, 35]. The loss of strength observed after curing at 320 oC may be related to violent dehydration cannot be tolerated by the permeability of the sample.
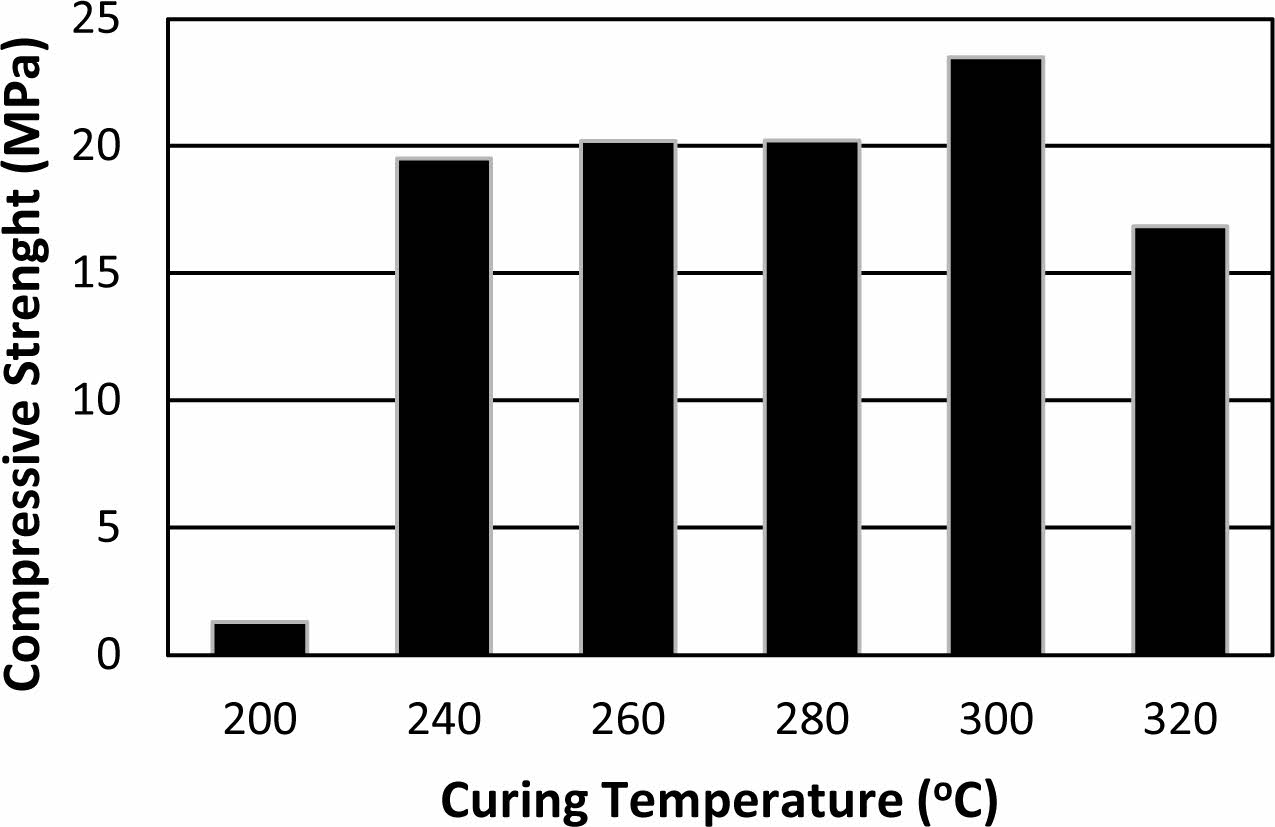
In Fig. 3, compressive strengths of the samples pro- duced at 300 oC with different binder addition ratios are given. Samples with 2.5 and 5 wt.% binder addition showed relatively low compressive strength such as 0.2 and 0.6 MPa, respectively. Steinerova investigated the different sand and binder ratios on mechanical and microstructural properties of metakaolin based geo- polymer composites [36]. Abundance of the binder and lack of binder both led to lower strength at final com- posites and optimum composition detected at 74-78 wt.% sand content [36]. Similar behaviour was observed for the binder produced in this study. It is thought that the lower compressive strength of the samples with a lower binder ratio than 20 wt.% is because the binder cannot cover the grain surfaces efficiently. Compressive strength increased gradually with increasing binder ratio up to 20 wt.% reaching 23.4 MPa before decrease again with increasing binder addition. Unlike the pro- duced binder, the abundance of the binder in formulations of metakaolin based geopolymer-sand composites lead to lower compressive strength but not lead to expansion due to lack of violent evaporation in their curing process. The curing technique applied in this study need to have enough permeability at the samples to allow dehydra- tion without deformation. This necessity limits the maximum dosage of the binder in formulations. The lower compressive strength of the samples with the higher amount of binder than 20 wt.% may be related to the reduced grain connectivity because of excessive binder and the deformation that occurred at the samples due to insufficient permeability.
Phase structure of the samples
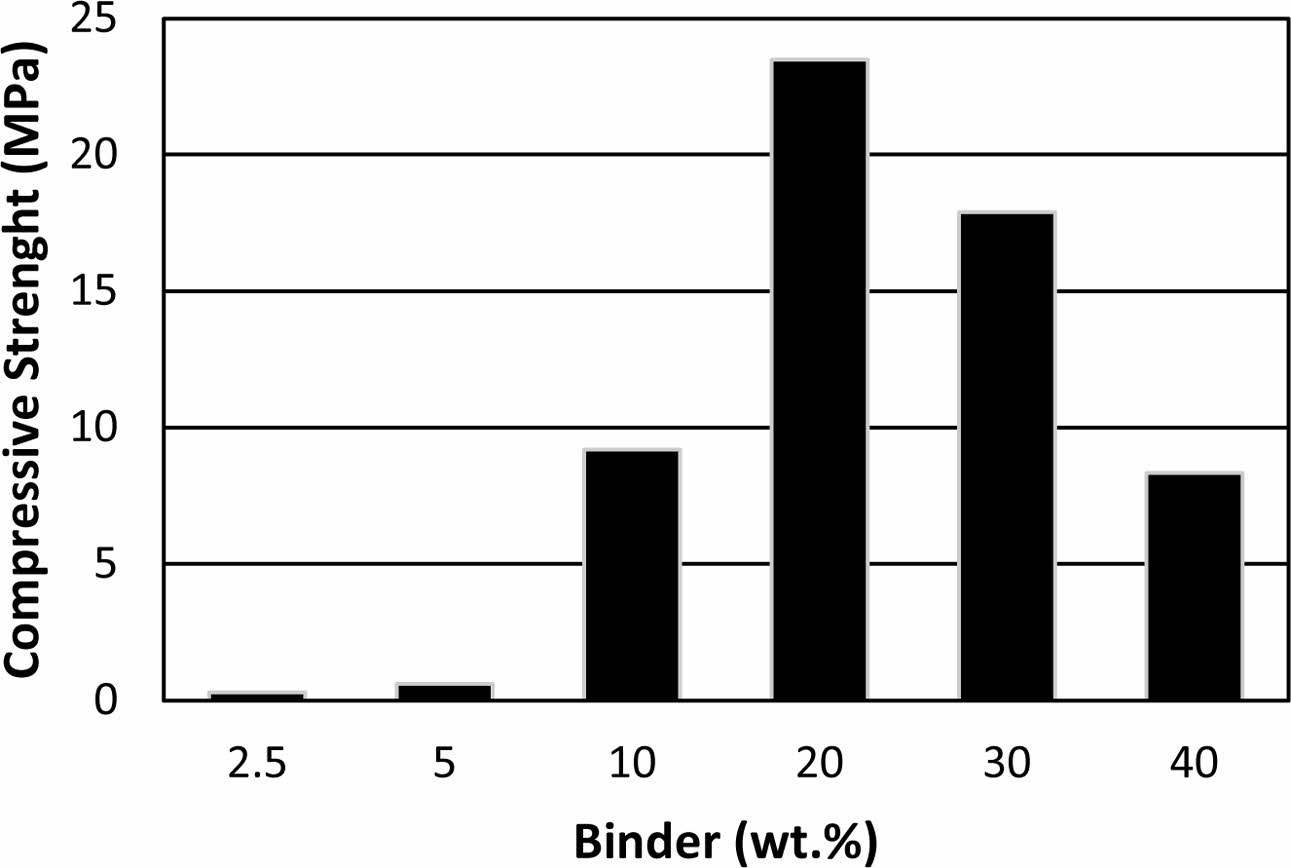
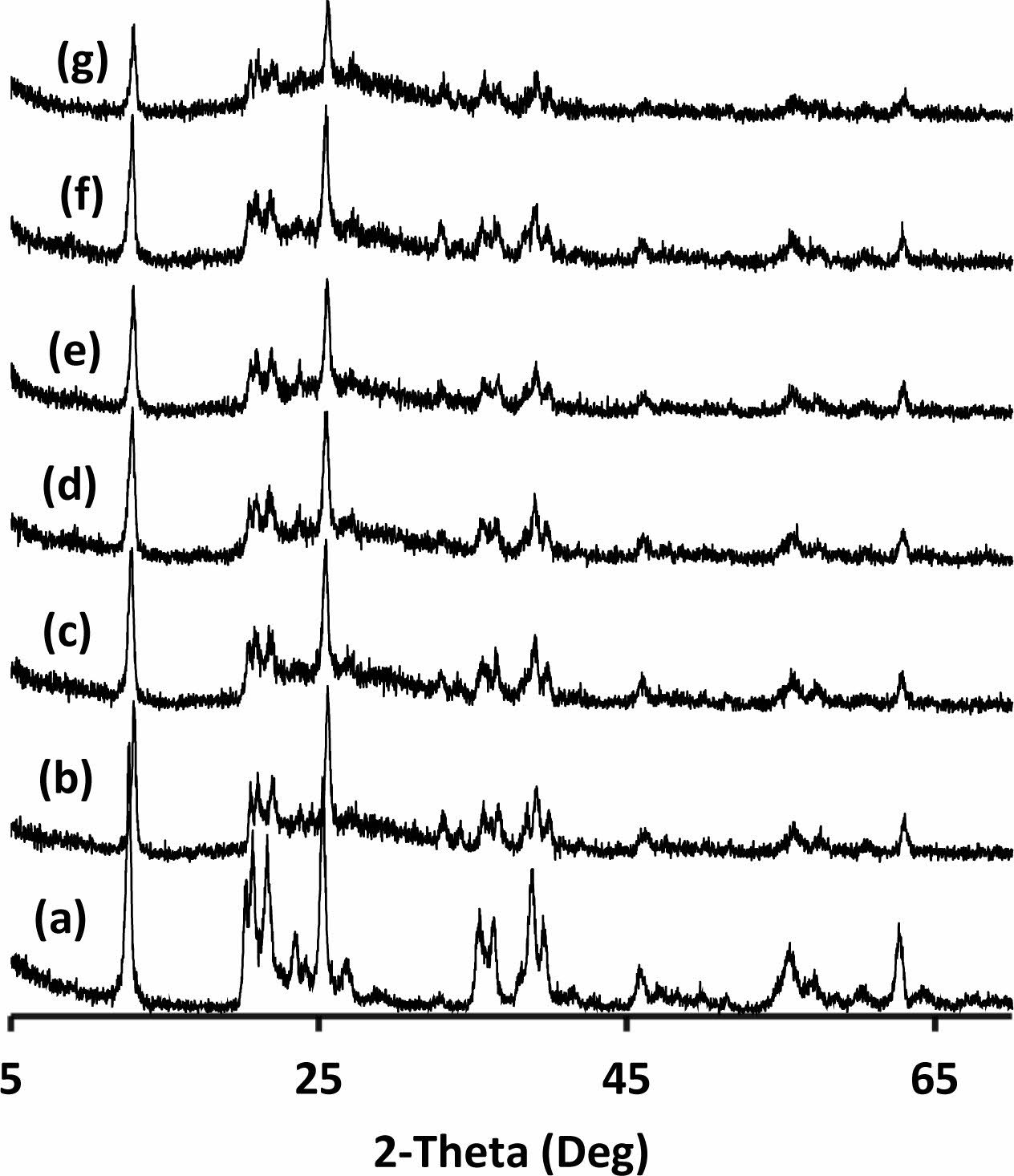
Only kaolinite peaks were detected for the kaolin powders (Fig. 4a). In addition to the crystal structure of the kaolin, an amorphous phase was seen in the binder (Fig. 4b-g). Heah et. al. reported that XRD analyses of kaolin based geopolymers activated by sodium silicate based solution show amorphous geopolymeric phase and crystalline peaks of unreacted part of the kaolin [15]. Hydrosodalite reported as a main crystalline product of the reactions between kaolin and NaOH solution for the samples cured in an air atmosphere at 80 oC for 24 h [37]. Zhang et al. investigated the effects of NaOH and sodium silicate solutions on crystal phase formation in metakaolin-based geopolymers [38]. They stated that crystalline phases occur in the metakaolin-based geo- polymer, which is only activated by NaOH, and the NaOH content significantly influences the nature, amount, and rate of crystallisation of the phases [38]. Even low amounts of sodium silicate addition suppressed the crystallisation [38]. The amorphous phase in the produced samples may be due to the use of sodium silicate solution in their formulations. Increased amorphousness can be attributed to increased geopolymerization because geo- polymeric phases were amorphous [1]. It was observed that amorphousness was increase and the intensity of kaolin peaks were decrease significantly in the sample exposed to 320 oC (Fig. 4g). This transformation may be a sign of more geopolymerisation of the hydrated alumina silicate precursor with increasing temperature in the studied range. Boutterin and Davidovits reported that the un-calcined kaolin based geopolymeric cross-linked bricks geopolymerised at 450 oC have signifi- cantly more compressive strength than bricks geopoly- merised at 85 oC [30]. This result supports the idea of more geopolymerisation of un-calcined kaolin with increasing temperature. Barbosa and MacKenzie reported that the geopolymeric phase remains amorphous up to 800 to 1,100 oC related to production parameters then crystallise to high-temperature phases [39]. Elimbi et. al. also reported that the crystalline phases appeared around 900 oC in the geopolymer samples [35]. Heat curing temperatures were not enough to lead to cry- stallisation of the amorphous geopolymeric phase in this study with increasing temperature so there was no crystallisation observed with increasing curing temper- ature.
FT-IR analyses of the samples
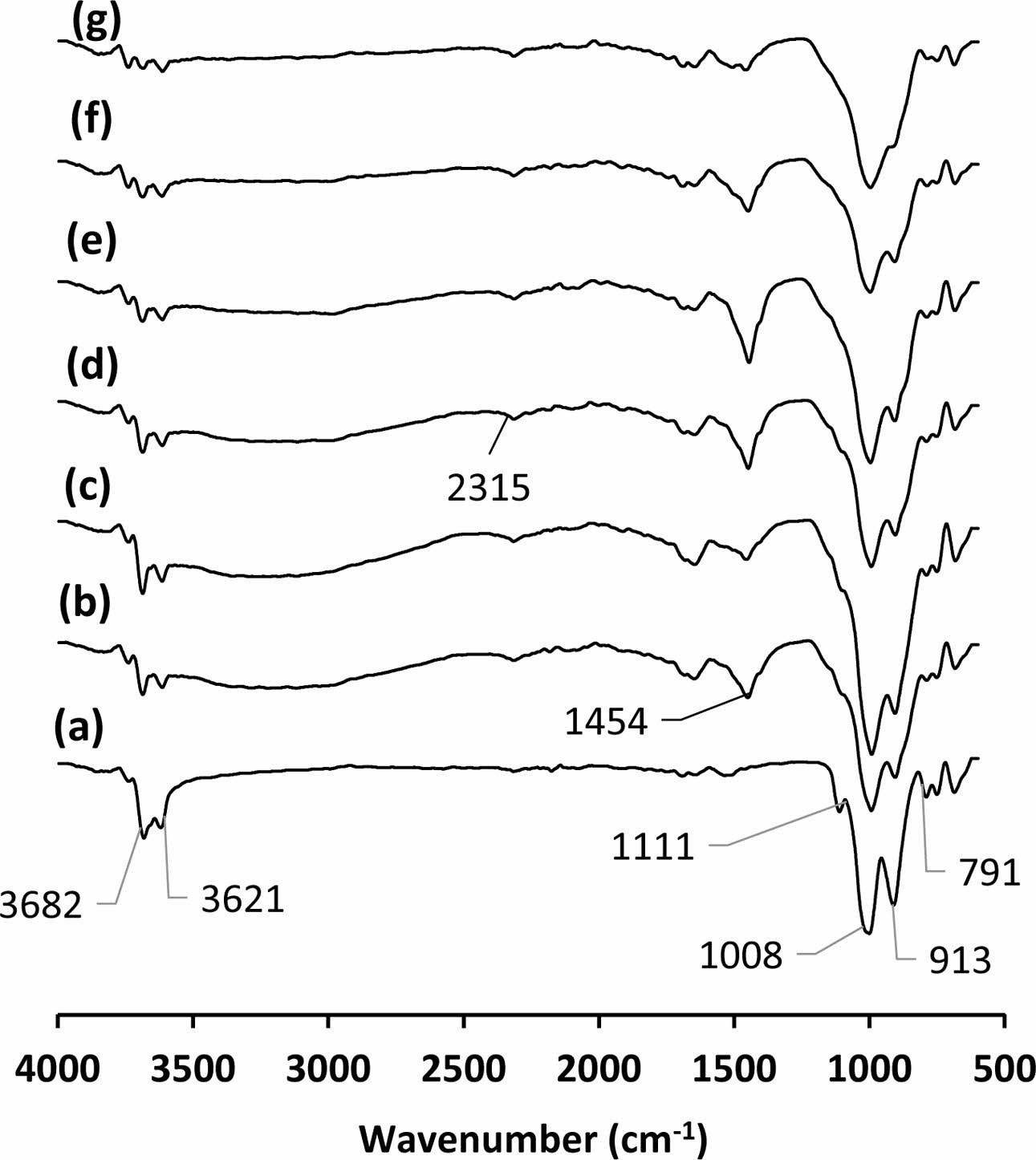
Peaks at the 791, 913, 1008 and 1111 cm-1 observed in the FTIR spectrum of kaolin (Fig. 5a) indicate Si-O, Al-OH, Si-O and Si-O bonds respectively [40, 41]. Peaks at 3621 and 3682 cm-1 originate from OH- molecules [41]. Atmospheric carbonation is commonly reported for alkali-activated materials [39, 42, 43]. It was also detected in the produced samples (Fig. 5b-g). The peaks around 1454 cm-1 results from the formation of Na2CO3 [43]. The peaks around 2315 cm-1 probably due to HCO3- ions [44]. Intensity of the peaks originating from OH- molecules around 3600 cm-1 in all samples reduced with increasing curing temperatures due to possible dehydroxylation or polycondensation reactions. The peak intensity of Al-OH bonds in kaolin at 913 cm-1 was reduced in the cured sample at 200 oC (Fig. 5a and b). The intensity of this peak gradually decreased with increasing curing temperature (Fig. 5b-g). The con- densation reactions releasing water molecule occurring between silanols of sodium silicate solution and hydro- xides of kaolinite mineral may consume this bond. As a result of this reaction, sialate (Si-O-Al-O) bridges are expected to form within the geopolymeric amorphous structure. Si-O-Al bonds in geopolymers are characterized by the presence of a wide peak between 900-1000 cm-1 wave number also observed in this study [41]. Yunsheng et al. reported that the reduced or disappeared peaks at 914 and 798 cm-1, belongs to stretching vibration of 6 fold coordinated Al(VI)-OH and 6 fold coordinated Al(VI)-O, respectively, in IR spectra of calcined kaolin after geopolymerisation means that 6-coordinated Al(VI) changed into 4-coordinated one at framework structure of geopolymer [40]. This finding regarding metakaolin and its geopolymerisation strengthens the idea of accepting the consuming of Al-OH- bonds with re- actions as evidence for the interaction between kaolin and sodium silicate solution in the samples. This reaction demonstrates the mechanism of kaolin as a geopoly- meric promoter for heat cured sodium silicate binder. It is reported that the un-calcined kaolin offers more strength than metakaolin with the heat curing method for foundry applications [27]. The abundance of OH- molecules in uncalcined kaolin provides a chemical advantage over calcined kaolin in terms of the reaction [27].
DTA-TG analyses of the samples
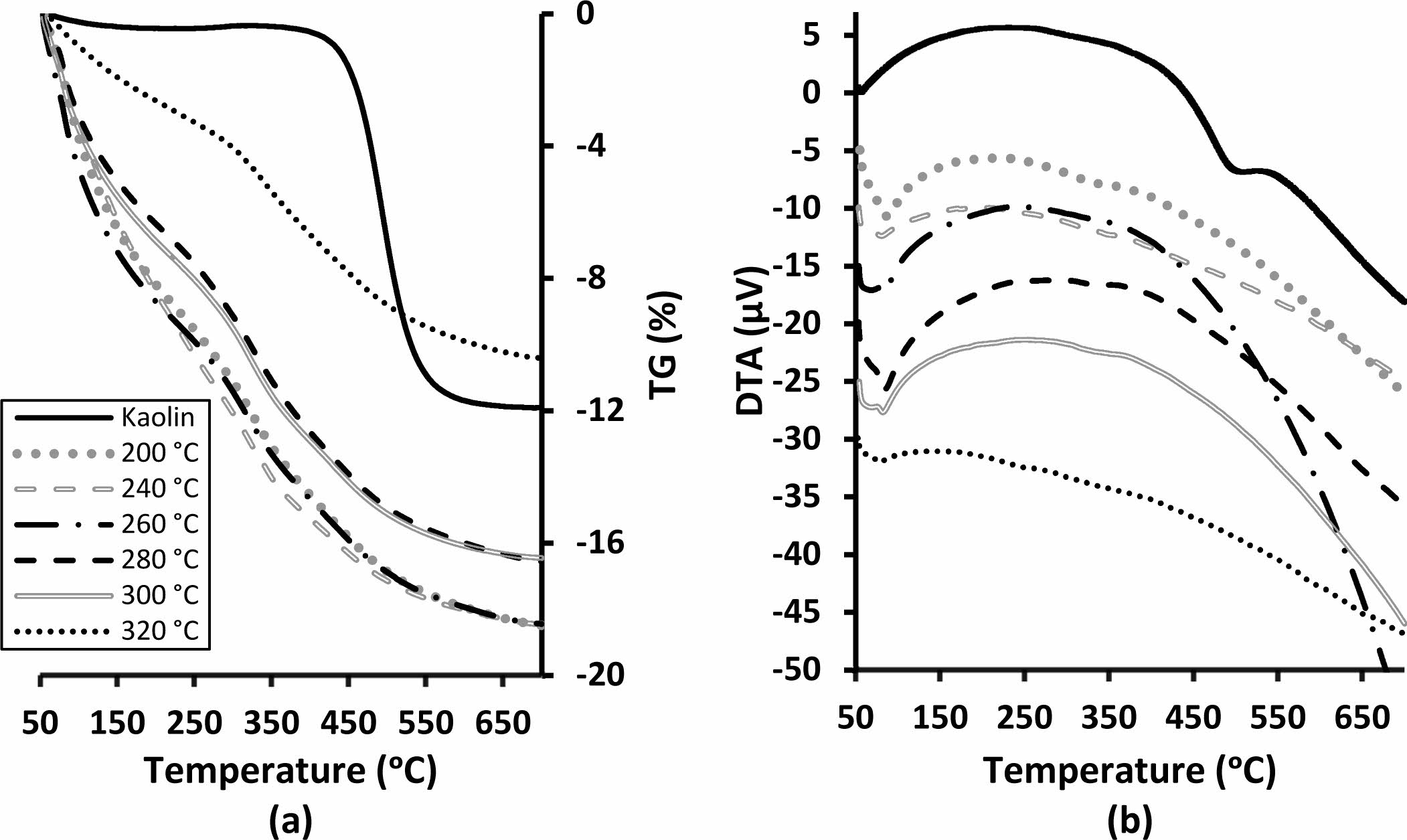
As a result of the characteristic endothermic dehydroxyl- ation reaction, 11.9 wt.% mass loss was observed from the kaolin (Fig. 6). Endothermic dehydration reactions at a temperature around 82 oC were observed in the DTA curve of all produced samples. (Fig. 6b). Total thermal mass losses of between 16-18 wt.% at 700 oC were observed for the binders cured up to 300 oC. (Fig. 6a). The binder cured at 320 oC, showed significantly lower thermal mass loss as 10 wt.% for the same temperature (Fig. 6a). Metakaolin-based geopolymers gradually lose mass over a wide temperature range due to dehydration and dehydroxylation [34, 35]. The binder produced in this study also showed similar thermal behaviour. However, despite the higher curing tempera- tures of the samples, the significant losses in mass at low temperatures indicate rehydration. Mass loss of the samples between room temperature and their curing temperature may be useful data to compare hygroscopic nature of the samples. The rehydration in the sample cured at 320 oC was significantly lower than the others (Fig. 6a). Decreasing water affinity is indicates that the condensation reactions continue with increasing tem- perature. It should be noted here that DTA-TG samples were produced without sand. The drying behaviour of the bulk materials with sand can vary depending on many physical factors such as binder dosage, temperature, duration, packaging density, size, and geometry. For example, the samples cured at 200 oC showed lower strength because of insufficient dehydration.
Microstructural analyses of the samples
The microstructures of the 20 wt.% binder added samples cured at different temperatures are given in Fig. 7. Loose and discontinuous microstructure was observed in the sample cured at 200 oC (Fig. 7a). The reasons may be the atmospheric moisture exposure with time and the insufficient reaction degree after heat curing. Samples had more compact microstructures when cured at 240 oC (Fig. 7b). The dramatic change in the microstructure was in good agreement with the compressive strength values.
SEM images of the samples cured at 300 oC with different binder additions are given in Fig. 8. Lack of binder in the samples observed by gaps between the sand for the addition ratios of 2.5, 5, and 10 wt.% (Fig. 8a-c). In the sample with 20 wt.% binder addition, the gaps between the grains were relatively filled by the binder, resulting in a denser structure (Fig. 8d). With 30 and 40 wt.% binder addition, it was observed that the gaps were almost filled. The connectivity of the grains decreased in proportion to the increasing binder amount up to 40 wt.% (Fig. 8e-f). This behaviour indicates the abundance of the binder was the reason of lower permeability in the samples produced with 30-40 wt.% binder addition.

|
Fig. 1 Appearance of the samples with (a) 2.5, (b) 5, (c) 10, (d) 20, (e) 30 and (f) 40 wt.% binder addition cured at 300 oC |
|
Fig. 2 Compressive strength of the samples with 20 wt.% binder addition cured at (a) 200, (b) 240, (c) 260, (d) 280, (e)300 and (f) 320 oC. |
|
Fig. 3 Compressive strength of the samples with (a) 2.5, (b) 5, (c) 10, (d) 20, (e) 30 and (f) 40 wt.% binder addition cured at 300 oC. |
|
Fig. 4 XRD pattern of the (a) kaolin and the binder cured at (b) 200, (c) 240, (d) 260, (e) 280, (f) 300 and (g)320 oC (all peaks belong to kaolinite at the pattern). |
|
Fig. 5 FTIR spectrum of the (a) kaolin and the binder cured at (b) 200, (c) 240, (d) 260, (e) 280, (f) 300 and (g) 320 oC |
|
Fig. 6 (a) TG and (b) DTA of the kaolin and the binders cured at various temperatures. |

|
Fig. 7 Microstructure of the samples with 20 wt.% binder addition cured at (a) 200 and (b) 240 oC. |

|
Fig. 8 Microstructure of the samples with (a) 2.5, (b) 5, (c) 10, (d) 20, (e) 30 and (f) 40 wt.% binder addition cured at 300 oC. |
Samples containing un-calcined kaolin and sodium silicate based geopolymeric binder and CEN standard sand were produced by heat curing up to 320 oC. The effects of the curing temperature and the additive ratio of the binder on the final product were examined. When the curing temperature reached 240 oC, a significant increase in the compressive strength and significantly denser microstructure of the samples were observed compared to samples cured at 200 oC. However, the highest compressive strength was obtained as 23.4 MPa in the sample cured at 300 oC. In the XRD analysis of the samples, the amorphous phase related to geopoly- merisation and kaolinite peaks that belongs to the un- reacted part of the precursor were detected. With in- creasing curing temperatures, the crystallinity decreased due to reactions between silanols of sodium silicate solution and hydroxides of kaolinite mineral to yield sialate (Si-O-Al-O) bridges that detected in the FT-IR spectra of the samples. In FT-IR analyses, the intensity of peaks indicating the Al-OH- bond originating from the kaolin decreased in proportion to the increasing curing temperature related to same reaction that consume hydroxides. The maximum binder loading limit was 20 wt.% to obtain deformation free products with the standard sand filler. The highest strength was also obtained at the same ratio. A higher amount of binder addition led to low permeability observed in the microstructures. To obtain deformation-free products designers should use the binder at the amount that allows dehydration from the sample without deformation during the heat curing.
We acknowledge the Karabük University, Turkey to support this project under the project code (KBÜBAP-18-YL-061). All analyses were performed in MARGEM Laboratory (KBU, Turkey).
- 1. J. Davidovits, J. Therm. Anal. 37 (1991) 1633-1656.
-
- 2. Q. Tian, D. Sun, Z. Gu and Z. Lv, J. Ceram. Process. Res. 21[3] (2020) 358-364.
-
- 3. J. Davidovits, in Proceedings of the Geopolymer 2002 Conference, October 2002, edited by G. Lukey (Siloxo Pty. Ltd. Press, 2002) p. 1.
- 4. C. Kuenzel, L.M. Grover, L.Vandeperre, A.R. Boccaccini and C.R. Cheeseman, J. Eur. Ceram. Soc. 33 (2013) 251-258.
-
- 5. S. Yaşın and H. Ahlatcı, J. Aust. Ceram. Soc. 55 (2019) 587-593.
-
- 6. Y. Rho and S. Kang, J. Ceram. Process. Res. 21[S1] (2020) 74-80.
-
- 7. Z. Chen and S. Lee, J. Ceram. Process. Res. 19[5] (2018) 419-423.
- 8. J. Davidovits, U.S. Patent No. US4349386A (1982).
- 9. J. Davidovits, J. Ceram. Sci. Technol. 8 (2017) 335-350.
-
- 10. M. Asadi, R. Naghizadeh, A. Nemati, K. Arzani and R. Nassiri, J. Ceram. Process. Res. 13[4] (2012) 425-428.
- 11. A. Elimbi, H.K. Tchakoute and D. Njopwouo, Constr. Build. Mater. 25 (2011) 2805-2812.
-
- 12. B.B. Kenne Diffo, A. Elimbi, M. Cyr, J. Dika Manga and H. Tchakoute Kouamo, J. Asian. Ceram. Soc. 3 (2015) 130-138.
-
- 13. V. Medri, S. Fabbri, J. Dedecek, Z. Sobalik, Z. Tvaruzkova and A. Vaccari, Appl. Clay. Sci. 50 (2010) 538-545.
-
- 14. J. Davidovits, Ceram. Eng. Sci. Proc. 38 (2018) 201-214.
- 15. C.Y. Heah, H. Kamarudin, A.M. Mustafa Al Bakri, M. Bnhussain, M. Luqman, I. Khairul Nizar, C.M. Ruzaidi and Y.M. Liew, Int. J. Miner. Metall. Mater. 20[3] (2013) 313-322.
-
- 16. F. Slaty, H. Khoury, J. Wastiels and H. Rahier, Appl. Clay. Sci. 75-76 (2013) 120-125.
-
- 17. A.D. Hounsi, G.L. Lecomte-Nana, G. Djétéli and P. Blanchart, Constr. Build. Mater. 42 (2013) 105-113.
-
- 18. P. Ghosh, H. Kumar and K. Biswas, Int. J. Geotech. Eng. 10 (2016) 377-386.
-
- 19. H. Roggendorf, D. Böschel and J. Trempler J. Non. Cryst. Solids. 293-295 (2001) 752-757.
-
- 20. L. Zaretskiy, Inter. Metalcast. 13 (2019) 58-73.
-
- 21. L. Chen, Z. Wang, Y. Wang and J. Feng, Materials 9 (2016) 767.
-
- 22. M. Arnoult, M. Perronnet, A. Autef and S. Rossignol, J. Non Cryst. Solids. 495 (2018) 59-66.
-
- 23. J. Davidovits, in Proceedings First International Conference on Alkaline Cements and Concretes, October 1994, edited by P.V. Krivenko (VIPOL Stock Company Press, 1994) p.131.
- 24. H.J. Zhuang, H.Y. Zhang and H. Xu, Procedia Eng. 210 (2017) 126-131.
-
- 25. A. Burian, M. Přerovská and B. Antošová Czech Patent No. CZ2016459A3 (2018).
- 26. M. Vykoukal, A. Burian and M. Přerovská, Arch. Foundry Eng. 19 (2019) 109-116.
-
- 27. H.J. Twardowska and H.J. Langer, World Patent No. WO1995026321A1 (1995).
- 28. S. Yaşın and S. Demir, in Proceedings International Iron and Steel Symposium (UDCS’21), April 2021, edited by Y. Sun, E. Çevik and M. Turan (Karabük University Press, 2021) p.154.
- 29. M. Davidovics and J. Davidovits, European Patent No. EP0101714B1 (1987).
- 30. C. Boutterin and J. Davidovits, in Geopolymer’88, June 1988, edited by J. Davidovits and J. Orlinski (Geopolymer Institute Press, 1988) p.79.
- 31. M.M. Davidovics, U.S. Patent No. US20130130886A1 (2013).
- 32. J. Davidovits, in Proceedings of the World Congress Geopolymer, July 2005, edited by J. Davidovits (Geopolymer Institute Press, 2005) p.9.
- 33. S. Lucas, M.T. Tognonvi, J.L. Gelet, J. Soro and S. Rossignol, J. Non. Cryst. Solids. 357 (2011) 1310-1318.
-
- 34. P. He, D. Jia, M. Wang and Y. Zhou, Ceram. Int. 37 (2011) 59-63.
-
- 35. A. Elimbi, H.K. Tchakoute, M. Kondoh and J. Dika Manga, Ceram. Int. 40 (2014) 4515-4520.
-
- 36. M. Steinerova, Ceram -Silikaty 55 (2011) 362-372.
- 37. A. Marsh, A. Heath, P. Patureau, M. Evernden and P. Walker, Microporous Mesoporous Mater. 264 (2018) 125-132.
-
- 38. B. Zhang, K.J.D. MacKenzie and I.W.M. Brown, J. Mater. Sci. 44 (2009) 4668-4676.
-
- 39. V.F.F. Barbosa and K.J.D. MacKenzie, Mater. Res. Bull. 38 (2003) 319-331.
-
- 40. Z. Yunsheng, S. Wei and L. Zongjin Appl. Clay. Sci. 47 (2010) 271-275.
-
- 41. Y.M. Liew, H. Kamarudin, A.M. Mustafa Al Bakri, M. Luqman, I. Khairul Nizar, C.M. Ruzaidi and C.Y. Heah, Constr. Build. Mater. 30 (2012) 794-802.
-
- 42. I. Lancellotti, M. Catauro, C. Ponzoni, F. Bollino, C. Leonelli, J. Solid State Chem. 200 (2013) 341-348.
-
- 43. V.F.F. Barbosa, K.J.D. MacKenzie and C. Thaumaturgo, Int. J. Inorg. Mater. 2 (2000) 309-317.
-
- 44. D. Zaharaki, K. Komnitsas and V. Perdikatsis, J. Mater. Sci. 45 (2010) 2715-2724.
-
 This Article
This Article
-
2022; 23(1): 41-47
Published on Feb 28, 2022
- 10.36410/jcpr.2022.23.1.41
- Received on Aug 5, 2021
- Revised on Nov 9, 2021
- Accepted on Nov 27, 2021
Services
- Abstract
introduction
experimental
results and discussion
conclusion
- Acknowledgements
- References
- Full Text PDF
Shared
Correspondence to
- Süleyman Yaşın
-
Karabük University, Department of Metallurgy and Materials Engineering, 78050, Karabük, Turkey
Tel : +90 444 04 78 - E-mail: syasin@karabuk.edu.tr
Clean-Energy Research Institute(CRI), Hanyang University, 222, Wangsimni-ro, Seongdong-gu, Seoul, 04763, Korea
E-mail: jcpr@hanyang.ac.kr